RhoA/ROCK通路調控小膠質細胞極化的研究進展
2022-11-11 03:41:18肖迎港
實用臨床醫藥雜志 2022年20期
肖迎港, 高 巨
(1. 揚州大學醫學院, 江蘇 揚州, 225009;2. 揚州大學附屬蘇北人民醫院 麻醉科, 江蘇 揚州, 225001)
腦卒中、腦創傷、膿毒癥腦病和阿爾茨海默病(AD)等疾病會造成神經元水腫、死亡,損害正常的腦功能[1-3]。小膠質細胞是機體主要免疫細胞,可激活為M1與M2表型,分別發揮增強和減弱炎癥反應的作用[4]。研究[5]發現,促進小膠質細胞極化為M2型有益于減輕腦損傷和恢復神經元功能,這讓調控小膠質細胞極化成為改善認知功能的新興領域。
Ras同源基因家族蛋白A(RhoA)/Rho相關卷曲螺旋蛋白激酶(ROCK)通路參與細胞的多種生理活動,包括細胞骨架重構、收縮、遷移、吞噬、黏附、應激纖維形成、炎癥反應和血管新生等,近年來發現其與小膠質細胞的極化也密切相關[6-7]。ROCK作為Ras同源家族蛋白的下游靶點,具有ROCK1與ROCK2亞型,但兩者對小膠質細胞極化可能產生不同作用[8]。闡明ROCK調控小膠質細胞極化的過程并分析相關差異,有助于深入了解小膠質細胞極化的分子機制,可為尋找更加有效的腦損傷治療新靶點提供方向和理論依據。
1 小膠質細胞概述
1.1 小膠質細胞與巨噬細胞的關系
巨噬細胞主要來源于卵黃囊和胎肝[9], 小膠質細胞作為定居中樞神經系統的巨噬細胞同樣來源于卵黃囊[10], 兩者都在免疫應答中發揮重要作用。小膠質細胞不僅具有巨噬細胞介導炎癥、免疫監視、極化和吞噬細胞碎片等功能和特征,還共同表達多種表面標志物,包括CD11b、F4/80和鈣離子接頭蛋白(Iba)-1等[11-13]。腦損傷發生時,血腦屏障破壞,巨噬細胞浸潤腦組織,此時會難以準確區分巨噬細胞和小膠質細胞,所以中樞神經系統疾病研究中常將小膠質細胞稱為小膠質細胞/巨噬細胞[4]。
1.2 小膠質細胞極化
靜息態小膠質細胞受外界刺激后激活,通過經典激活途徑和替代激活途徑分別極化為M1與M2這2種功能迥異的細胞表型[5]。M1型小膠質細胞表達CD32、CD86和CD16等表面標記物,并分泌腫瘤壞死因子(TNF) -α、誘導型一氧化氮合酶(iNOS)、白細胞介素(IL)-1β和IL-6等促炎因子,加劇炎癥反應; M2型小膠質細胞則表達CD206和CD163等細胞表面標記物,分泌轉化生長因子(TGF)-β、精氨酸酶(Arg)-1、IL-4和IL-10等抑炎因子,吞噬細胞碎片,從而抑制炎癥反應并促進組織修復[5, 14-15]。小膠質細胞極化表型主要受機體內環境影響,組織液中脂多糖(LPS)和干擾素(INF)-γ能誘導小膠質細胞向M1型極化,而IL-10和IL-4可刺激M2型極化發生[16]。小膠質細胞極化后的產物能促使細胞繼續向初始極化方向改變,該過程是典型的正反饋調節。此外,間充質干細胞也會影響小膠質細胞極化[17]。M2型小膠質細胞存在多種亞型,包括M2a、M2b和M2c, M2a通過釋放抑炎因子和神經營養因子修復受損組織; M2b主要由Toll樣受體(TLR)/IL-18介導,表達抗炎介質; M2c具有吞噬功能,能清除細胞碎片,但無法殺滅病原菌[18]。
然而, M2型小膠質細胞也存在負面影響。研究[19]發現,在特發性肺纖維化患者中, M2型巨噬細胞表達增加會導致肺纖維化加重。這可能與M2型細胞釋放TGF-β、促進組織修復進而導致成纖維細胞增生有關。在眼科疾病研究中亦存在類似發現,脈絡膜中M2型小膠質細胞和巨噬細胞會導致血管增生,加重視力受損程度[8, 14]。此外,研究[20]發現M2型巨噬細胞也會促進高侵襲性口腔鱗癌細胞遷移。另有研究[21]表明,巨噬細胞經LPS刺激后雖然為M1型,釋放促炎因子,但產生的外泌體卻能促進小膠質細胞的M2型極化,這可能與外泌體中所含的微小RNA(miRNA)有關。上述結果表明, M2型小膠質細胞雖然發揮抗炎、促進損傷組織修復等積極作用,但其促進修復的作用如果過強或作用于癌細胞時,就會對機體產生嚴重后果。
2 RhoA/ROCK信號通路概述
2.1 RhoA家族
Rho鳥苷三磷酸酶包括3類分子: Rho、Rac、Cdc42。作為其中之一的Rho分子又細分為A、B、C、D、E共5種亞型,均能作用于下游ROCK分子。但RhoE較特殊,對ROCK起著抑制作用[22]。Rho蛋白作為小G蛋白的一種,同樣受鳥嘌呤核苷酸交換因子(GEFs)調控,靜息狀態下, Rho結合GDP, 無生物活性,但經GEFs催化后, Rho轉而結合GTP,轉化成激活態[23]。空間上, RhoA激活時會從胞質轉移到細胞膜,調節肌球蛋白收縮、產生應力纖維、形成黏著斑和偽足[24-25]。
2.2 ROCK1與ROCK2的異同
ROCK是RhoA的下游分子,存在ROCK1亞型與ROCK2亞型,二者結構極為相似,激酶結構域的同源性高達92%[26]。ROCK在全身組織中均有分布, ROCK1主要表達于非神經組織,如肝、肺和血液中, ROCK2則在腦和肌肉組織中占主導地位[27]。ROCK激活后磷酸化下游分子,主要包括肌球蛋白輕鏈(MLC)、肌球蛋白磷酸酯酶靶點亞單位(MYPT)1和肌球蛋白輕鏈磷酸酶(MLCP)[8]。磷酸化MYPT1會促進MLC磷酸化,而磷酸化的MLCP會喪失促進MLC脫磷酸基的作用[28]。兩者從增加來源和抑制去路2個方面調節磷酸化MLC含量。RhoA/ROCK通路主要通過增加磷酸化的MLC, 進而發揮多種基于細胞骨架改變的生理作用,如細胞間黏附[29]、細胞運動[30]、軸突回縮[31]和平滑肌細胞收縮[32]等,所以磷酸化MLC表達量在研究[27, 33]中常被用于判斷RhoA/ROCK信號通路激活程度。
RhoA/ROCK改變細胞骨架的機制主要與肌絲滑行有關。細胞膜受到刺激后,開放陽離子鈣通道,內流的鈣離子結合并激活肌鈣蛋白,帶動原肌球蛋白轉位,暴露出肌動蛋白。此后肌動蛋白再與肌球蛋白橫橋結合,引發肌細胞收縮或微絲滑動,改變細胞骨架。RhoA/ROCK通過作用于MLC參與這一過程,激活的ROCK使肌球蛋白輕鏈磷酸化,增加對肌動蛋白的敏感性,促進肌細胞收縮或改變細胞骨架[22, 32]。
3 RhoA/ROCK調控小膠質細胞極化
3.1 ROCK促進小膠質細胞M1型極化
細胞實驗[34]表明,對小膠質細胞進行氧糖剝奪/再灌注(OGD/R)[35]和LPS誘導,觸發了IL-1β和iNOS的釋放,以及相應M2型小膠質細胞的標志物下調,如Arg-1和CD206, 而對ROCK進行抑制后,上述導致損傷加重的分子則被有效逆轉。多項實驗[36-38]同樣證實了該結論,在慢性偏頭痛、實驗性過敏性腦脊髓炎、AD和腦缺血再灌注損傷小鼠模型中,RhoA/ROCK的表達都發生了上調,且iNOS、IL-6和CD86等M1型小膠質細胞的標志物表達增加。ROCK抑制劑則能有效抑制促炎因子釋放,增加抑炎因子釋放,減輕中樞神經系統損傷,改善認知功能。
雖然諸多研究表明抑制RhoA/ROCK后,能降低神經細胞死亡率,減輕腦損傷,但ROCK抑制劑的腦保護作用是否主要依靠小膠質細胞M2型極化,迄今尚不清楚。究其原因,炎癥下ROCK激活還會引發腦血管痙攣,增加血管通透性,破壞血腦屏障,這些均是加重腦損傷的重要因素。2014年的1項研究[31]為解決該問題提供了依據。研究人員用1-甲基-4-苯基-1, 2, 3, 6-四氫吡啶(MPTP)處理大鼠胚胎來源的中腦細胞培養液和不含小膠質細胞的中腦培養液,誘導其多巴胺能神經元變性,再對2種培養液使用ROCK抑制劑Y-27632干預,最后發現ROCK抑制劑可顯著減輕含小膠質細胞的中腦培養液中多巴胺能神經元變性死亡,在不含小膠質細胞的中腦培養液中卻未觀察到該現象。此體外細胞實驗表明,M2型小膠質細胞數量增加在ROCK抑制劑減輕神經元損傷的機制中發揮重要作用,但缺乏進一步動物實驗提供更有力的證據。
3.2 ROCK調控小膠質細胞極化的相關機制
3.2.1 神經元損傷時RhoA/ROCK引起的病理生理改變: 靜息態小膠質細胞胞體較小,具有纖細的分支狀結構,從而監控大腦微環境[40]。人罹患神經系統疾病或損傷導致內環境劇烈改變時,小膠質細胞隨之激活,胞體增大,突起回縮甚至消失,呈阿米巴樣,此時細胞代謝、運動和吞噬能力均會增強[8, 40-41], 且M2型小膠質細胞的吞噬能力強于M1型。而小膠質細胞激活時,這一系列形態改變均依賴于細胞骨架的可塑性, RhoA/ROCK通路的重要性不言而喻。RhoA/ROCK作為調控細胞骨架主要的信號通路參與其中。炎癥反應發生時,激活的RhoA/ROCK信號通路會減少肺血管內皮細胞間的緊密連接蛋白,增大毛細血管通透性,導致炎癥因子和免疫細胞滲出[42]。進入腦組織液中的炎癥因子,如IL-1、IL-10和TNF-α, 能直接促進小膠質細胞極化。將內皮剝脫后的大鼠主動脈環浸泡于含有GTP類似物GTPγS的溶液中,發現RhoA/ROCK通路激活,導致血管平滑肌收縮[33]。這提示發生腦損傷時,激活的ROCK可能使腦血管痙攣,加重腦缺血缺氧,引發更多炎癥因子聚集,導致定居于中樞神經系統的小膠質細胞和浸潤巨噬細胞持續極化。極化過程中RhoA/ROCK不僅是多種上游分子的靶點,還可以通過調控相關信號通路分子表達,進而調節小膠質細胞極化(見圖1)。
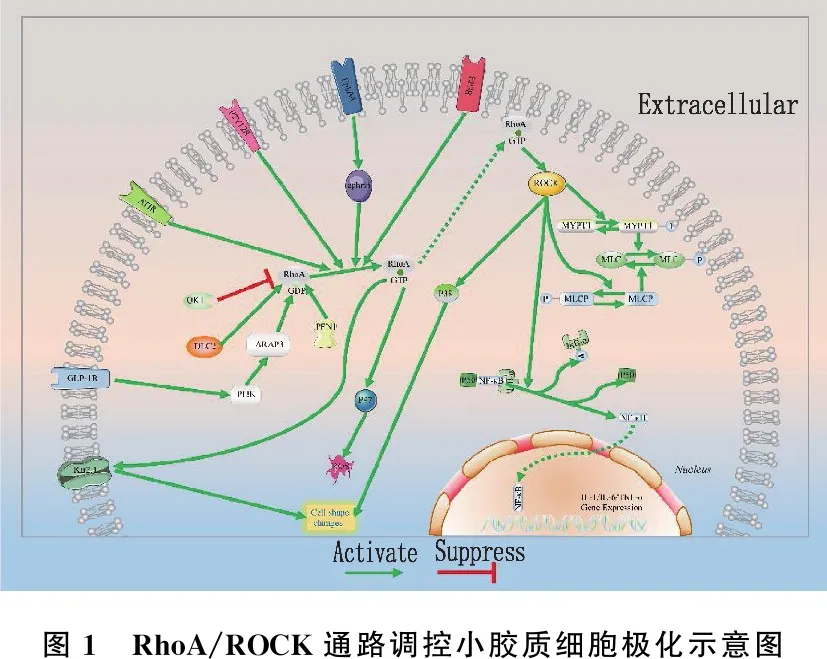
圖1 RhoA/ROCK通路調控小膠質細胞極化示意圖
3.2.2 極化過程中調節RhoA/ROCK的上游分子: 用硝酸甘油建立小鼠慢性偏頭痛(CM)模型, JING F等[36]發現模型中GTP-RhoA、ROCK2、降鈣素基因相關肽(CGRP)、c-fos和iNOS蛋白水平顯著升高; 進一步研究發現,嘌呤能受體P2Y12(P2Y12R)和ROCK2活性降低能抑制硝酸甘油(NTG)誘導的小膠質細胞形態改變并產生iNOS, 最后證明三叉神經尾側核中小膠質細胞P2Y12R激活后,通過RhoA/ROCK途徑調節小膠質細胞活化,且其在CM發病機制中起重要作用。此外,神經退行性疾病領域研究[1, 38, 43]發現, AD小鼠中淀粉樣蛋白沉淀會激活RhoA/ROCK通路,從而促進小膠質細胞的遷移、趨化、浸潤和吞噬。GUO M F等[1]進一步揭示了具體機制,法舒地爾作為廣泛認可并使用的ROCK抑制劑,在AD大鼠中發揮了抑制(TLR4/髓樣分化因子88(Myd88)/核因子-κB(NF-κB)通路的作用,促進小膠質細胞向M2型極化,增加M2型小膠質細胞的數量,但研究人員未討論RhoA/ROCK通路的作用。ROCK能直接促進NF-κB[44], 因此RhoA很可能是TLR4或Mydd88的下游靶點。而法舒地爾抑制TLR4的作用可能是由于分泌促炎因子的M1型小膠質細胞數量減少,而間接抑制TLR4相關通路。
另有研究[34]表明,阻斷神經元和小膠質細胞之間EphA4/ephrin信號通路后, RhoA/ROCK2通路被抑制,促進小膠質細胞M2極化,進而減輕OGD/R誘導的神經元凋亡。此外, RNA結合蛋白QUAKING(QKI)和肝癌細胞中缺失的抑癌基因DLC2分別抑制和上調RhoA表達,發揮小膠質細胞調控作用,影響神經細胞修復[45-46]。
除信號分子外,一些膜受體也在小膠質細胞極化過程中參與調控RhoA/ROCK。MPTP誘導的帕金森病模型[47]發現表達上調的血管緊張素(AT)Ⅱ結合AT受體后,激活RhoA/ROCK通路,從而增強小膠質細胞反應性并加重多巴胺能神經元變性。HAN X N等[48]發現凝血酶結合前列腺素E2的EP3受體, EP3受體偶聯RhoA/ROCK通路并將其激活,進而增加CD68陽性的M1型小膠質細胞,最終導致神經損傷。創傷性腦損傷(TBI)大鼠的傳統中醫治療[49]發現,手針能通過抑制RhoA/ROCK2通路抑制小膠質細胞M1型極化,減輕急性顱腦創傷后的神經炎癥。這一結論提示RhoA/ROCK通路可能受到機械門控通道的調節。此外,腸促胰島素類似物-4(EX-4)能激活胰高血糖素樣肽-1受體(GLP-1R), 從而在轉錄水平抑制磷脂酰肌醇3-激酶(PI3K)/ARAP3/RhoA通路,促進小膠質細胞M2極化,減輕神經損傷[50]。
3.2.3 極化過程中由RhoA/ROCK影響的下游分子: RhoA/ROCK在調節極化過程中還涉及諸多其他中間信號分子,如激發炎癥級聯反應的NF-κB[44]。NF-κB 作為被廣泛研究的重要轉錄因子,通常以p50-NF-κB二聚體形式與NF-κB抑制蛋白-α(IκB-α)結合,在細胞質中處于非活性狀態; 當受相關因子刺激時, IκB-α磷酸化降解, NF-κB與p50分離并迅速發生轉核,結合調控基因啟動子上的κB位點,進而啟動IL-1、IL-6和TNF-α等促炎因子基因轉錄。RhoA/ROCK磷酸化下游靶目標IκB-α, IκB-α隨后從與NF-κB組成的復合體中解離,不再抑制NF-κB進入細胞核發揮作用[8]。
小鼠腦缺血再灌注模型中, LU E M等[39]發現,血清前纖維蛋白1(PFN1)下調能減少p38絲裂原活化蛋白激酶(p38MAPK)表達并且抑制RhoA/ROCK激活,導致M2型極化。進一步研究發現, p38MAPK在福爾馬林引起炎性疼痛小鼠模型[45]和脊髓損傷大鼠模型[51]中受RhoA/ROCK通路激活,促進小膠質細胞聚集。對脊髓損傷大鼠模型其他相關研究[52]還發現, p38MAPK在RhoA/ROCK調節小膠質細胞形態變化中也起關鍵作用。但目前仍缺乏強有力證據證明p38MAPK與M1型極化直接相關。除了最關鍵的p65NF-κB和p38MAPK因子, MUESSEL M J 等[53]發現Kir2.1鉀通道也是RhoA引起小膠質細胞形態變化的重要中間分子。RhoA還能通過磷酸化煙酰胺腺嘌呤二核苷酸磷酸(NADPH)氧化酶亞基p47, 促進M1型BV2細胞(小鼠小膠質細胞)分泌活性氧(ROS)[54]。
RhoA/ROCK通路對小膠質細胞極化的調控是多方面的,既有改變細胞骨架,促進細胞形態變化的直接調節,又有通過激活其他信號通路,引發炎癥因子聚集,對小膠質細胞的間接活化。
3.3 ROCK與小膠質細胞極化關系的探討
多數觀點認為抑制RhoA/ROCK活化能通過促進小膠質細胞M2型極化產生有益作用,但有部分研究得出了不同結論。采用熒光免疫方法分析脈絡膜新生血管(CNV)小鼠和猴脈絡膜巨噬細胞,發現RhoA/ROCK表達上調導致M2型巨噬細胞增多,進一步研究[8]發現激活ROCK1促進M1型極化,而ROCK2促進M2型細胞增多。基于該結論,有研究[14]發現,對CNV小鼠注射褪黑素能顯著抑制眼部炎癥導致的RhoA/ROCK通路激活,進而抑制小膠質細胞M2型極化。LPS使BV-2細胞向M1型極化,而藍莓提取物促進細胞骨架相關Rho蛋白表達,進而逆轉小膠質細胞極化方向[55]。
雖然有研究證實RhoA/ROCK既能促進M1型極化,又能促進M2型極化,但是RhoA/ROCK本身就在炎性微環境中激活,且組織損傷導致免疫細胞分泌炎癥因子的過程也依賴細胞骨架改變。此外,ROCK還能以增大血管通透性、收縮血管等方式加重炎癥反應,所以RhoA/ROCK信號通路激活更可能促進小膠質細胞M1型極化。細胞不管是何種極化,都會導致細胞形態變化與細胞因子分泌,而分泌細胞因子必然借助于囊泡形成。這均涉及細胞骨架改變,因此作為改變細胞骨架的關鍵通路RhoA/ROCK很可能參與其中。M1型與M2型細胞中上調的RhoA/ROCK分子使MLC蛋白磷酸化從而改變細胞骨架,但M1型中RhoA/ROCL分子的另一核心作用是激活NF-κB和MAPK這2種關鍵且廣泛的促炎分子,在胞核中從基因轉錄水平影響小膠質細胞極化過程。因此, RhoA/ROCK通路更傾向介導M1型極化,但也參與M2型極化。抑制RhoA/ROCK導致M2型細胞增加,可能是將過度上調的RhoA/ROCK降至主要改變M2型細胞骨架的水平。此時,小膠質細胞極化更傾向于M2型。
4 結 語
綜上所述, RhoA/ROCK通路在小膠質細胞作用發揮的調控中具有重要地位,其上下游存在廣泛的中間因子,抑制其激活促使小膠質細胞從加重炎癥的M1型極化,向具有神經保護作用的M2型轉化。雖然ROCK調節小膠質細胞極化的作用被廣泛研究,但因缺乏選擇性抑制ROCK1或ROCK2的藥物,所以臨床針對ROCK分子的靶向治療陷入了瓶頸。此外, ROCK分子在中樞神經系統損傷時作用廣泛,其中復雜的關聯機制亟待進一步研究闡明。RhoA/ROCK分子分布于全身各組織細胞,而外泌體技術可能在輔助ROCK抑制劑穿過血腦屏障、靶向作用于中樞神經系統的過程中發揮重要作用。ROCK抑制劑法舒地爾因具有改善血管痙攣的作用,成為臨床上治療蛛網膜下腔出血的常用藥物,但其介導小膠質細胞極化產生抗炎作用的研究多集中在基礎實驗。KOCH J C等[56]報道3例肌萎縮側索硬化癥患者經法舒地爾治療后,病情得到顯著改善,且無明顯副作用,這可能與其調控極化的作用有關。未來更多的相關臨床研究有望推動擴大RhoA/ROCK抑制劑的適用范圍。
盡管存在諸多難題,但隨著研究手段不斷發展,各種分子如miRNA、長鏈非編碼RNA(lncRNA)和外泌體對RhoA/ROCK極化調控機制進一步闡明, RhoA/ROCK通路相關藥物及診療手段有望在小膠質細胞與腦保護領域中發揮更有意義的作用。
