高效液相色譜法測定醋酸來法莫林的有關物質
2022-11-11 12:04:16何美翠張春然黃清東王宇馳王瑛瑛于瑤張靜霞李知儀趙歡唐克慧
中國抗生素雜志 2022年9期
何美翠 張春然,* 黃清東 王宇馳 王瑛瑛 于瑤 張靜霞 李知儀 趙歡 唐克慧
(1 抗生素研究與再評價四川省重點實驗室,四川抗菌素工業研究所,藥學院,成都大學,成都 610106;2 四川百特文理科技有限公司,成都 611731)
醋酸來法莫林(lefamulin acetate)為來法莫林的原料藥,化學名為14-O-{[(1R,2R,4R)-4-氨基-2-羥基環己基硫烷基]乙酰基}-截短側耳素乙酸鹽[1](圖1)是第一個系統性給藥的截短側耳素類抗生素,為Nabriva公司開發研制。2019年8月19日成功在美國獲批,后又相繼在歐盟和加拿大獲批,用于治療成人社區獲得性細菌性肺炎(CABP)[2]。商品名:Xenleta?,作為近20年來FDA批準的第一款具有獨特作用機制的抗生素,其同時擁有兩種劑型(靜脈注射和口服),在常見和難治性感染均具潛在療效,未來具有廣闊的應用空間[3]。醋酸來法莫林作為截短側耳素藥物具有獨特的作用機制,與當前抑制細菌蛋白質合成的抗生素均不易產生交叉耐藥性,因而在治療社區獲得性細菌性肺炎具有不可替代的作用。目前,各國藥典及標準均未收載該藥與其制劑的質量標準,同時醋酸來法莫林有關物質檢測的方法未見文獻報道。本文參考截短側耳素類藥物的分析方法,建立了適用于醋酸來法莫林有關物質測定的高效液相色譜法。
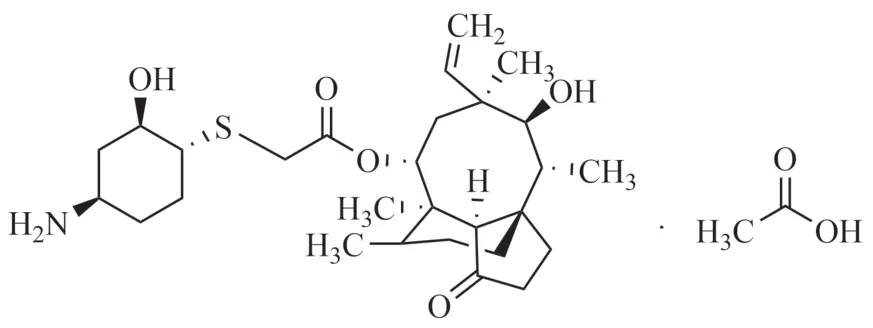
圖1 醋酸來法莫林的結構式Fig.1 Structural formula of lefamulin acetate
1 儀器與試藥
Shimadzu LC-20AB高效液相色譜儀;Agilent 1200 Serial液相色譜儀;島津UV-2450紫外分光光度計;Sartorius BP211D型天平。
醋酸來法莫林粗品(批號:20200527)、樣品(批號:20200915、20200929、20201029);起始原料Ⅰ(截短側耳素),起始原料Ⅱ((R)-3環己烯甲酸);中間體Ⅰ(S-((1R,2R,4R)-2-hydroxy-4-(2,2,2-trifluoroacetamido)cyclohexyl)benzothioate),中間體Ⅱ(來法莫林);試劑(硫代苯甲酸)。以上試藥試劑均由四川百特文理科技有限公司提供;乙腈為HPLC級(美國Fisher);水為純凈水(娃哈哈);其余試劑均為分析純。
2 方法與結果
2.1 色譜條件
色譜柱:中譜紅PR-C18色譜柱(25 cm×4.6 mm,5 μm),流動相A:60 mmol/L磷酸二氫鉀溶液(磷酸調pH至2.5)-乙腈(80:20,V/V),流動相B:60 mmol/L磷酸二氫鉀溶液(磷酸調pH至2.5)-乙腈(50:50,V/V),梯度洗脫(表1),流速1 mL/min,檢測波長為210 nm,柱溫為30℃,進樣體積為20 μL。

表1 梯度程序Tab.1 Gradient elution program
2.2 溶液配制
溶劑:乙腈:水(2:8,V/V)
供試品溶液:取醋酸來法莫林適量,精密稱定,加溶劑溶解并稀釋制成每1 mL中約含5 mg的溶液。
對照溶液:精密量取供試品溶液適量,加溶劑稀釋制成每1 mL中含50 μg的溶液。
系統適用性溶液:精密稱取醋酸來法莫林和起始原料Ⅱ各適量,加溶劑溶解并稀釋制成每1 mL中各約含0.5 mg的溶液。
靈敏度溶液:取系統適用性溶液適量,加溶劑稀釋制成每1 mL中各約含2.5 μg的溶液。
2.3 有關物質測定
系統適用性要求:系統適用性溶液色譜圖(圖2)中,理論板數按來法莫林峰計算不得低于5000,來法莫林峰與起始原料Ⅱ的分離度大于1.5。靈敏度溶液色譜圖(圖2)中,主成分峰高的信噪比應不低于10。
測定法:以“2.1”項下的液相色譜方法,按“2.2”項配制供試品溶液和對照溶液,精密量取對照溶液20 μL注入液相色譜儀,調節檢測靈敏度,使主成分色譜峰的峰高約為滿量程的10%~20%;再精密量取供試品溶液和對照溶液各20 μL注入液相色譜儀,記錄色譜圖。供試品溶液的液相色譜圖(圖2)中如顯示雜質峰,均按不加校正因子的主成分自身對照法進行計算。
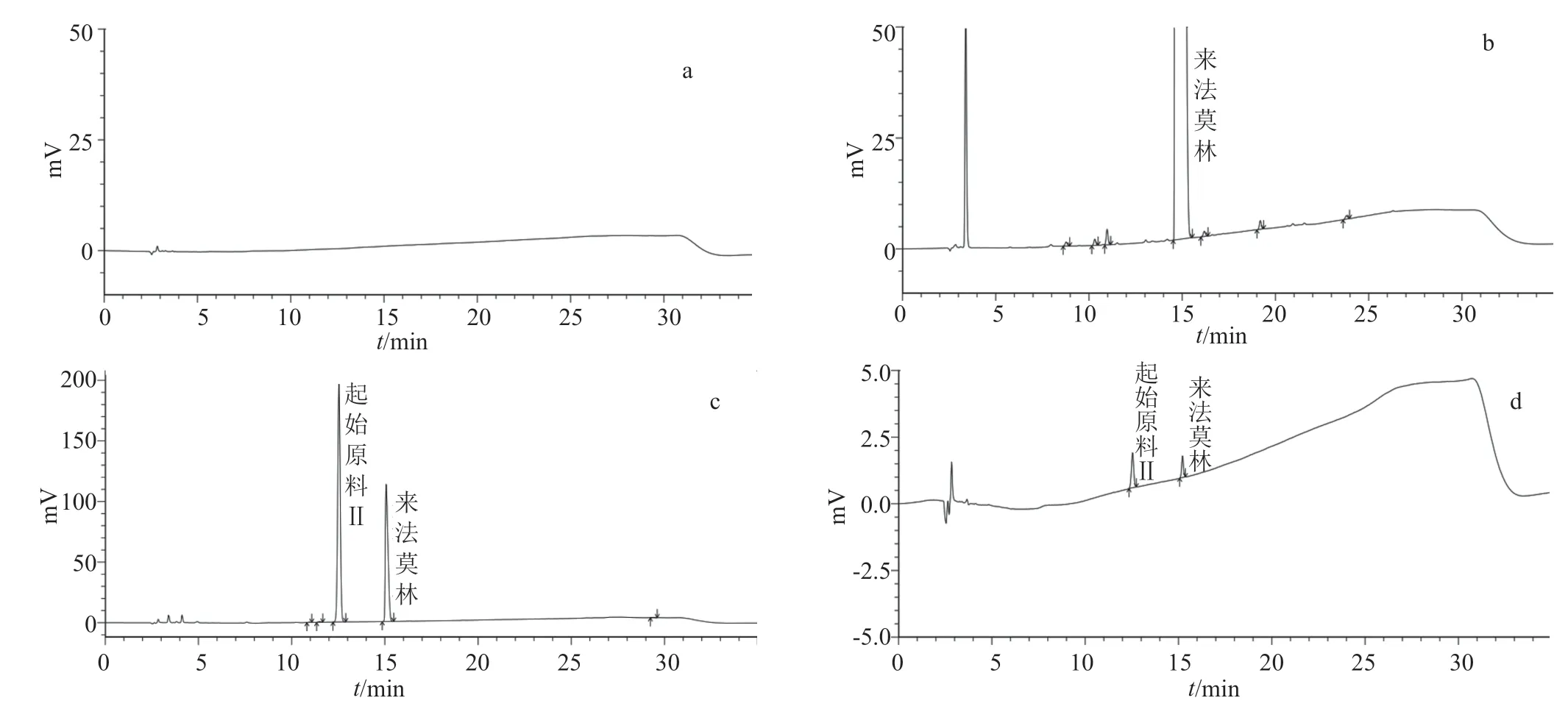
圖2 醋酸來法莫林有關物質測定相關HPLC圖Fig.2 Related HPLC chart for determination of related substances in lefamulin acetate
2.4 方法學驗證
2.4.1 專屬性考察
(1)與原料、中間體、試劑的分離情況
取醋酸來法莫林樣品(批號:20200915,方法學驗證均用該批次)適量,再加入適量起始原料、中間體、試劑,加20%乙腈(起始原料或中間體溶解度差時可加乙腈溶解后,再加20%乙腈稀釋至刻度)溶解并稀釋制成每1 mL中約含5 mg醋酸來法莫林、25 μg來法莫林、10 μg中間體Ⅰ、5 μg硫代苯甲酸、25 μg(R)-3環己烯甲酸、50 μg的截短側耳素的混合溶液。
(2)與降解雜質的分離情況
酸破壞:精密稱取醋酸來法莫林25 mg于5 mL容量瓶中,以1 mL 4 mol/L HCl溶液水浴60℃條件下破壞樣品1 h,于該酸破壞溶液加入1 mL 4 mol/L NaOH溶液中和,用1 mL乙腈溶解樣品,再以20%乙腈稀釋至刻度。
堿破壞:精密稱取醋酸來法莫林25 mg于5 mL容量瓶中,以1 mL 1mol/L NaOH溶液水浴60℃條件下破壞樣品1 h,于該堿破壞溶液加入1 mL 1 mol/L HCl溶液中和,加1 mL乙腈溶解樣品,再以20%乙腈稀釋至刻度。
氧化破壞:精密稱取醋酸來法莫林25 mg于5 mL容量瓶中,以1 mL 3% H2O2溶液于室溫下破壞樣品30 min,再以20%乙腈稀釋至刻度。
高溫破壞:取醋酸來法莫林適量,在高溫(120℃)條件下放置4 h,取出后于干燥器中冷卻至室溫,精密稱取樣品適量,制成5 mg/mL的溶液。
高濕破壞:取醋酸來法莫林適量,于高濕環境(25℃,相對濕度(RH)為75%±5%)中放置10 d,精密稱取樣品適量,制成5 mg/mL的溶液。
光照破壞:取醋酸來法莫林樣品適量,于ChP2020四部通則9001規定的光照強度(4500±500 Lx)下放置10 d,精密稱取樣品適量,制成5 mg/mL的溶液。
精密量取醋酸來法莫林供試品溶液以及上述6種破壞溶液各20 μL,按“2.1”項下的色譜條件分別進樣檢測并記錄色譜圖。相應圖譜見圖3,由試驗結果可知,醋酸來法莫林于光照破壞、高濕破壞條件下較穩定,而在其余4種破壞條件下均會發生降解,其中氧化降解產生兩個雜質(氧化降解雜質A和氧化降解雜質B,以下稱雜質A和雜質B),在上述的色譜條件下,起始原料、中間體及各降解產物均能與醋酸來法莫林主峰完全分離,證明所建方法專屬性強。
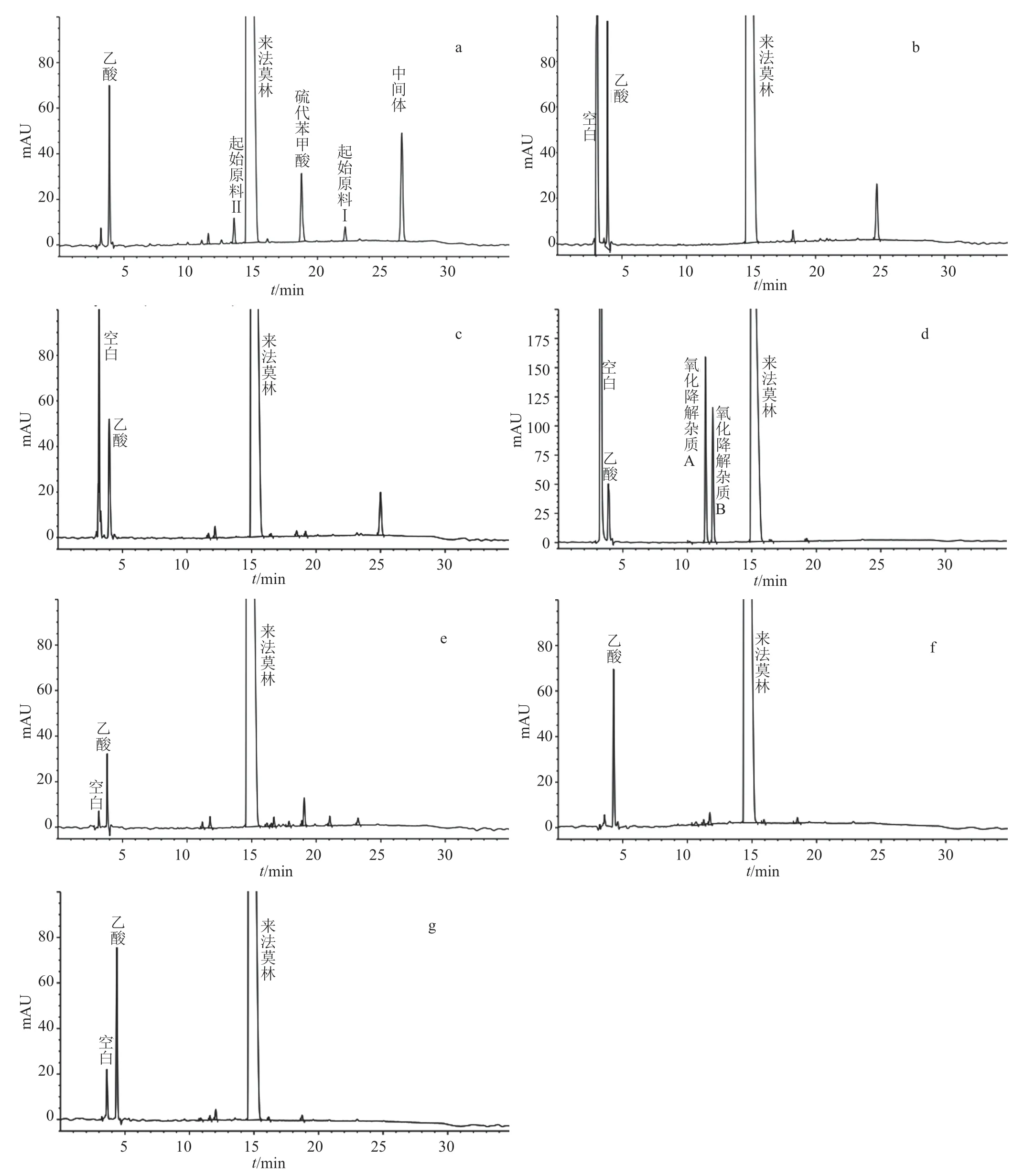
圖3 專屬性試驗HPLC色譜圖Fig.3 The HPLC chromatogram of specificity test
2.4.2 線性
精密稱取醋酸來法莫林樣品適量于10 mL容量瓶,加20%乙腈溶解并稀釋制成每1 mL約含5 mg的儲備液。精密量取醋酸來法莫林儲備液適量分別制成濃度為0.50、1.00、5.02、12.56、25.11、50.22和75.33 μg/mL的醋酸來法莫林溶液,按“2.1”項下液相色譜條件進樣分析并記錄譜圖。以主峰峰面積(y)對醋酸來法莫林濃度(x)進行線性回歸,回歸方程:y=2113.4x-173.23,R2(相關系數)為1.000。由結果可知,醋酸來法莫林在0.50~75.33 μg/mL濃度范圍內與峰面積呈較好的線性關系。
2.4.3 檢測限和定量限
取醋酸來法莫林線性溶液逐步稀釋,根據島津LC solution QAQC計算操作步驟來計算信噪比(S/N),以信噪比S/N=15.62對應的醋酸來法莫林濃度0.50 μg/mL為定量限,以信噪比S/N=7.73對應的醋酸來法莫林濃度0.20 μg/mL為檢測限。
2.4.4 供試品溶液的穩定性試驗
取醋酸來法莫林一定量,按“2.2”項制備供試品溶液,于室溫放置0、1、2、4、6、8、10、12和24 h,按“2.1”項下色譜條件進樣檢測并記錄譜圖,供試溶液主峰峰面積RSD(相對標準偏差)值為0.31%,雜質A的RSD為0,雜質B含量的RSD值為1.44%,總雜質含量的RSD值為1.86%,表明醋酸來法莫林溶液在24 h內穩定性良好。
2.4.5 重復性試驗
精密稱取醋酸來法莫林樣品6份,依照“2.2”項制備溶液,按照“2.1”項下色譜條件進樣檢測并記錄譜圖,以醋酸來法莫林樣品中雜質A、雜質B、最大單個未知雜質及總雜質的含量考察方法的精密度,雜質A的RSD值為0,雜質B的RSD值為1.74%,最大單個未知雜質的RSD值為0,總雜質的RSD值為1.39%。
2.4.6 中間精密度
以同一批醋酸來法莫林樣品,按照“2.2”項制備溶液,在不同時間由不同分析人員分別在Shimadzu LC-20AB和Agilent 1200 Series儀器上按“2.1”項下的色譜條件進樣檢測,以醋酸來法莫林樣品中的雜質A、雜質B、最大單個未知雜質及總雜質含量考察方法的中間精密度,雜質A的RSD值為0,雜質B的RSD值為1.99%,最大單個未知雜質RSD值為0,總雜質RSD值為1.47%。結果證明所建方法精密度較好。
2.4.7 耐用性試驗
依照“2.2”項制備醋酸來法莫林供試品及對照溶液,按“2.1”項下的色譜條件進樣檢測,分別考察流速、柱溫、波長等條件有微小變化時,醋酸來法莫林的色譜行為,結果見表2。由表可知,樣品測定條件有小的變動時,各項測定結果、雜質檢出個數相同,證明所建方法耐用性較好。
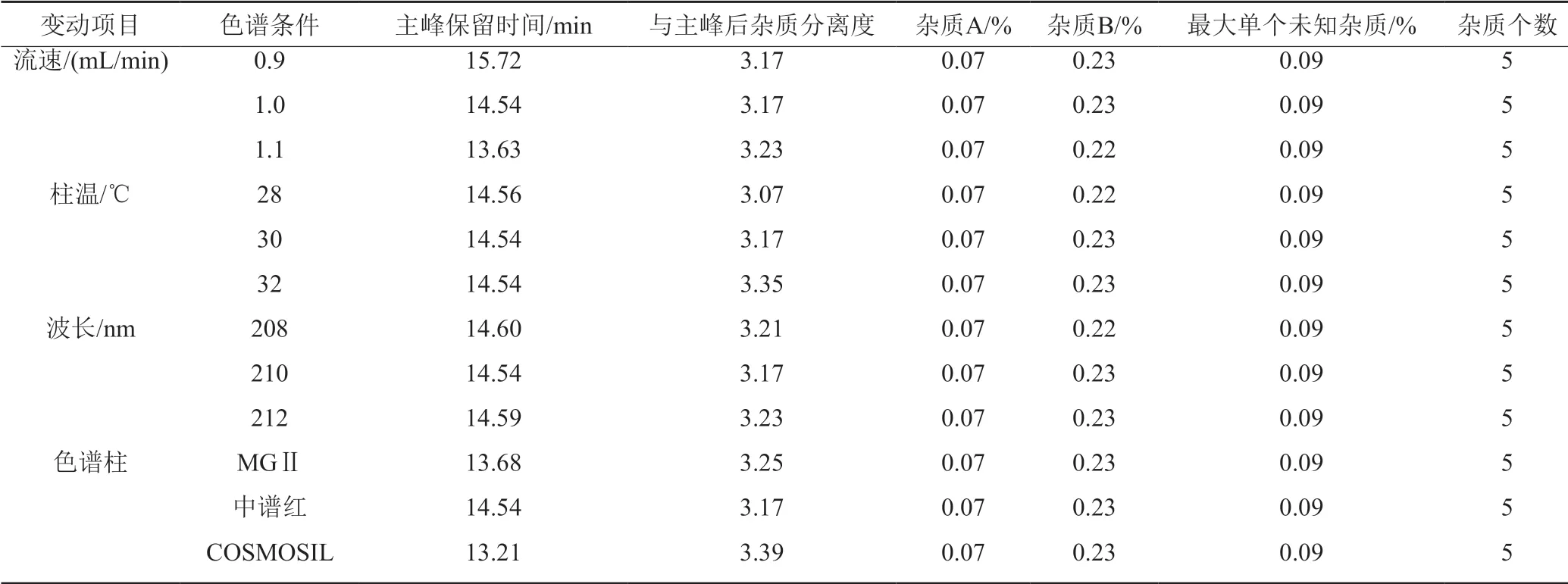
表2 耐用性試驗結果Tab.2 Durability test results
2.5 有關物質檢查結果
按照“2.2”項制備醋酸來法莫林供試品及其對照溶液,按“2.1”項下的色譜條件進樣分析,對3批醋酸來法莫林樣品有關物質進行檢查,檢查結果見表3。根據結果可知,3批醋酸來法莫林樣品的雜質A、雜質B、最大單個未知雜質含量、總雜質含量差別不大。

表3 醋酸來法莫林有關物質檢查結果Tab.3 Test results of related substances of lefamulin acetate
3 討論
3.1 色譜條件的選擇與確定
洗脫程序的確定:參考截短側耳素類藥物的分析方法[4],以磷酸鹽-乙腈為流動相,等度洗脫,雜質與主峰未能分開;梯度洗脫,雜質與主峰能有效分離,調節梯度程序使各雜質與主成分的分離度提高。水相的確定:①磷酸二氫鈉和磷酸氫二鈉、②磷酸氫二鉀與磷酸二氫鉀、③磷酸氫二鈉和磷酸二氫鉀、④磷酸二氫鉀、⑤磷酸氫二鈉、⑥0.1%磷酸、⑦0.1%TFA(三氟乙酸)、⑧0.1%甲酸,發現水相中的鹽離子可以使主峰的峰形收斂,不同的緩沖鹽作為水相,檢出雜質個數、相鄰雜質之間的分離度均相差不大,使用鉀鹽比鈉鹽的基線平穩。綜合考慮后,選用磷酸二氫鉀作為緩沖鹽。
3.2 檢測波長的選擇
取醋酸來法莫林粗品(批號:20200527)適量,精密稱定,以水為稀釋劑,制成1 mg/mL的溶液,在200~800 nm波長范圍內進行光譜掃描,結果顯示,醋酸來法莫林在203和244 nm處有最大吸收峰,在400~800 nm波長范圍內無吸收。由于醋酸來法莫林樣品243 nm波長處的吸收較小,雜質檢出數目少;將檢測波長改為210 nm時的吸收增大,且210 nm波長下檢出的雜質較多,故我們初步將210 nm作為檢測波長。采用二極管陣列檢測器,對樣品、破壞樣品進行全波長掃描,所得圖譜見圖4;結果顯示,在210 nm波長處,主峰、雜質峰的純度因子均大于計算的閾值,表明各峰均為單峰,同時,在其他波長處未顯示新的吸收峰,說明無雜質漏檢。因此,選定能檢出雜質最多、主峰和雜質峰均有較大吸收的210 nm作為檢測波長。
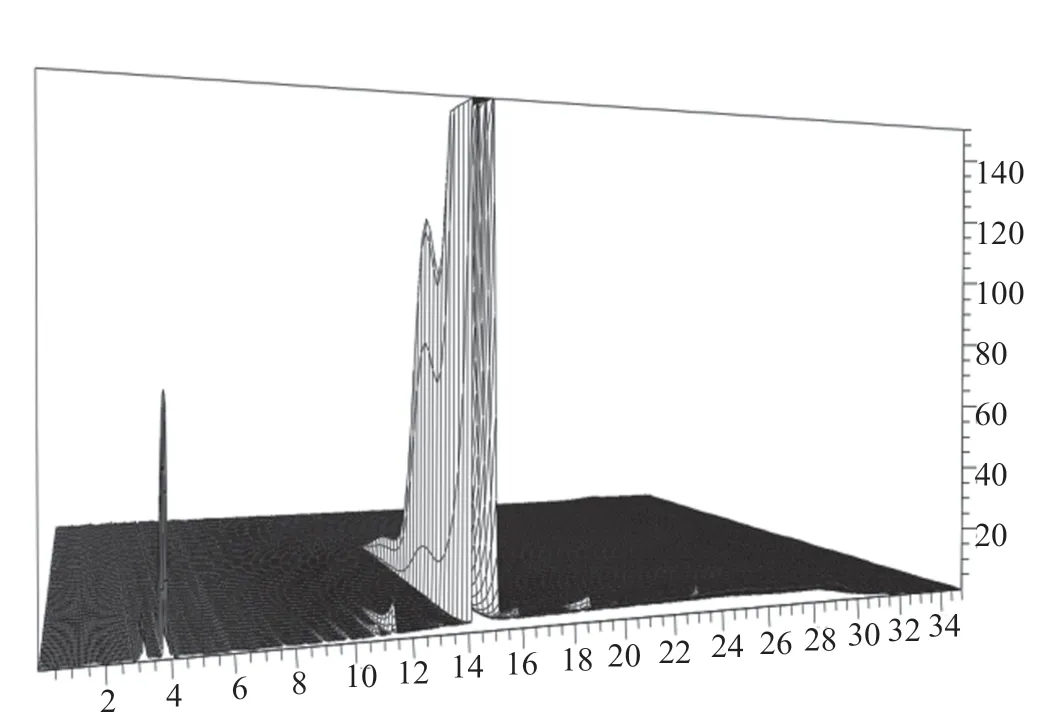
圖4 醋酸來法莫林的二極管陣列掃描圖Fig.4 The diode array scannogram of lefamulin acetate
3.3 磷酸二氫鉀溶液pH的選擇
流動相中磷酸二氫鉀溶液的pH為2.0、2.5、3.0、4.5和5.5,考察磷酸二氫鉀溶液pH的變化對醋酸來法莫林(批號:20200527)主峰保留時間、主峰理論板數、主峰與鄰近雜質峰分離度及雜質檢出個數的影響。結果表明:pH5.5時,基線漂移嚴重。pH在2.0至4.5范圍內隨著緩沖鹽pH值增加,檢出雜質個數由8個變為7個,而主峰柱效、拖尾因子、主峰與鄰近雜質峰之間的分離度均差異不大。綜合考慮后選擇pH為2.5。
3.4 磷酸二氫鉀溶液濃度的選擇
流動相中磷酸二氫鉀的濃度分別為10、25、50、60、68和75 mmol/L,考察磷酸二氫鉀濃度的變化對醋酸來法莫林(批號:20200527)主峰保留時間、主峰理論塔板數、拖尾因子及主峰與相鄰雜質峰分離度的影響。結果表明,隨著磷酸二氫鉀溶液濃度的增加,拖尾因子降低,理論塔板數升高、主峰與相鄰雜質峰分離度增加。當流動相中緩沖鹽濃度達到60 mmol/L時,繼續增加磷酸二氫鉀溶液濃度,雖主峰理論板數稍有增加,但拖尾因子、主峰與鄰近雜質峰分離度兩個指標變化不大。綜合考慮,將緩沖鹽濃度定為60 mmol/L。
3.5 進樣濃度的選擇
取醋酸來法莫林(批號:20200527)適量,分別配制成濃度為1.0、2.5、5.0、6.0、7.0、8.0和10.0 mg/mL的溶液,同法進樣分析。結果顯示,溶液濃度為5 mg/mL時,再繼續增加樣品濃度,檢出的雜質個數不變,故將醋酸來法莫林有關物質檢查濃度定為5 mg/mL。
3.6 雜質A、B分析
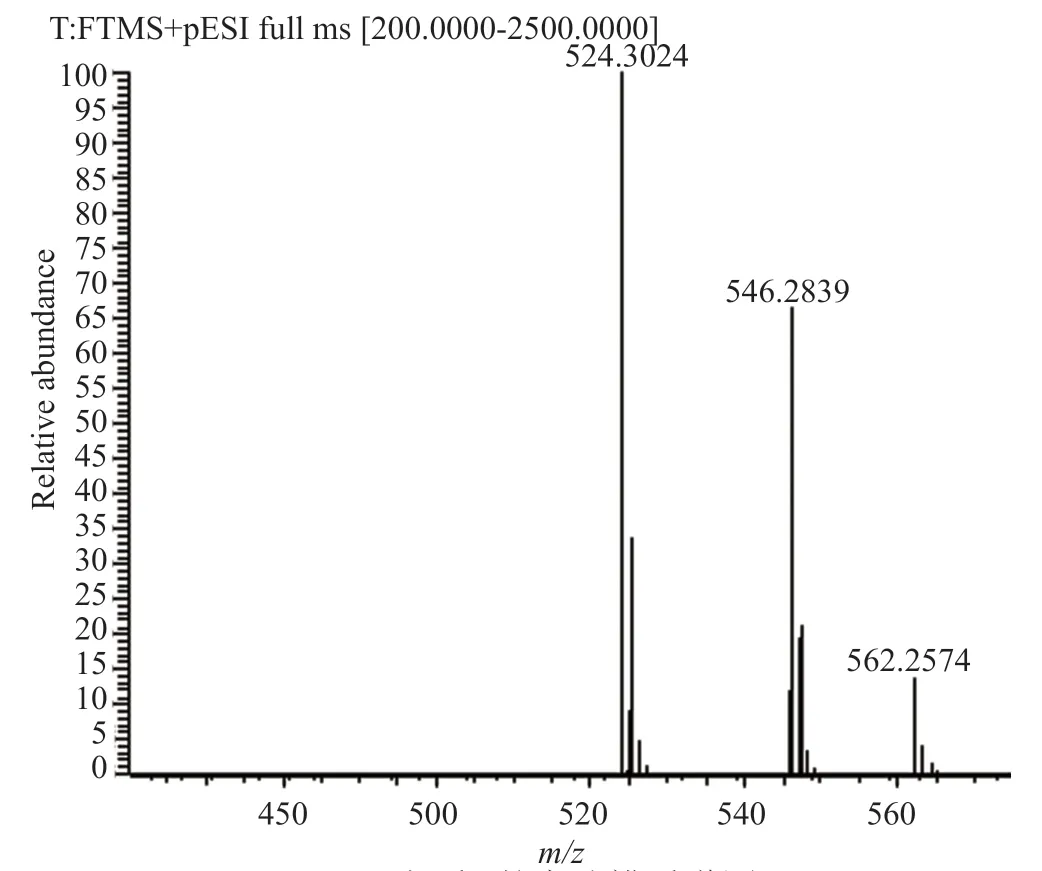
圖6 雜質B的高分辨質譜圖Fig.6 HR-MS chart of impurity B
在樣品中,雜質B的歸一化含量大于0.1%,根據ICH的文件Q3A和中國藥典四部通則藥品雜質分析指導原則,需要對該雜質進行研究。首先采用起始原料與中間體定位雜質,結果表明雜質A、B既不是原料也不是中間體;進而采用最后一步反應剩余的母液分析雜質來源,也未能成功定位歸一化含量大于0.1%的雜質;最后根據降解試驗產生的降解雜質分析雜質來源,其中氧化降解產生2個較大的雜質,兩雜質與樣品中歸一化含量較大及該雜質前一相鄰雜質保留時間一致。通過半制備高效液相色譜法收集雜質A溶液與雜質B溶液,再將所得的兩溶液進行高分辨質譜分析,兩雜質的高分辨質譜圖見圖5~6。兩雜質的ESI正離子譜圖顯示其分子離子峰有m/z524.3023、546.2839、562.2574,分別對應[M+H]+、[M+Na]+、[M+K]+離子峰,表明其分子量為523,而來法莫林的分子量為507,氧化降解產生的兩雜質剛好比來法莫林多了一個氧原子的分子量16,研究證明雜質A、雜質B為氧化降解雜質,結合醋酸來法莫林的結構與合成路線,推測雜質A、B可能為醋酸來法莫林硫醚鍵氧化成亞砜或碳碳雙鍵環氧化的產物,結構見圖7。在氧化劑(過氧化氫)存在的條件下,醋酸來法莫林結構中的硫醚鍵會被氧化成亞砜,碳碳雙鍵則被氧化成環氧乙烷。結合醋酸來法莫林的合成路線分析樣品中雜質A與雜質B的來源,推測是空氣中的氧氣將硫醚氧化成亞砜,有研究表明[5]在沒有氧化劑的條件下可以利用氧氣將硫醚氧化成亞砜,因空氣中氧氣占比較大,可能在反應過程中將硫醚氧化成亞砜。同理,碳碳雙鍵環氧化也是空氣中氧氣的作用[6]。
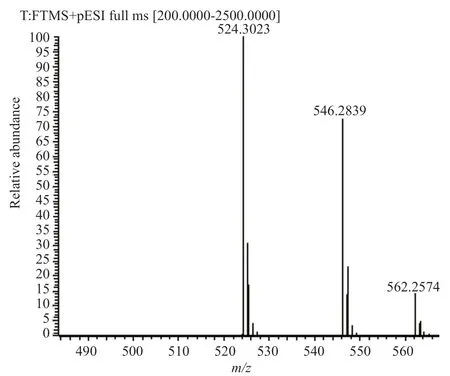
圖5 雜質A的高分辨質譜圖Fig.5 HR-MS chart of impurity A
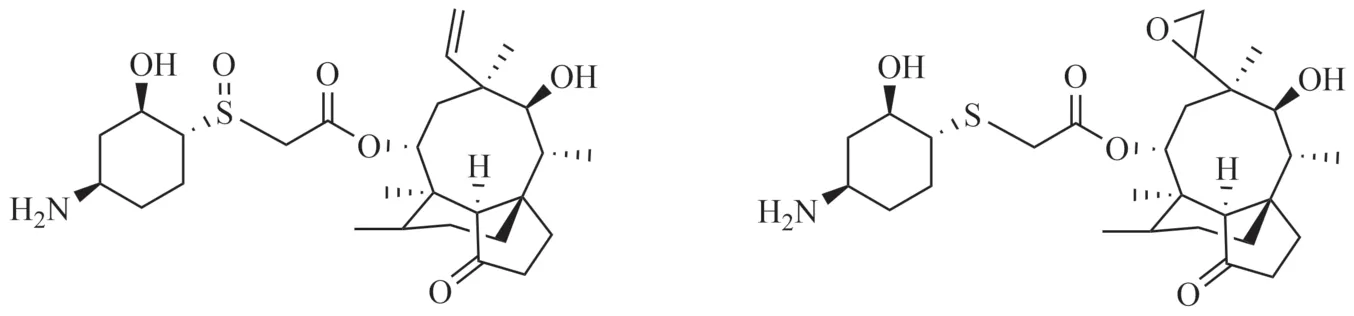
圖7 雜質A或雜質B可能的結構Fig.7 Possible structure of impurity A or impurity B
