納米制劑克服腫瘤多藥耐藥的研究進展
2022-11-11 12:58:58匡正秦超楊磊尹莉芳韓曉鵬
藥學研究 2022年10期
關鍵詞:耐藥
匡正,秦超,楊磊,尹莉芳,韓曉鵬
(中國藥科大學藥學院,江蘇 南京 211198)
腫瘤的多藥耐藥性(multidrug resistance,MDR)是導致癌癥化療失敗的重要原因之一,也是臨床癌癥治療的最大挑戰[1]。MDR指長期接觸某一化療藥物而產生的不僅對此種化療藥物耐藥性,而且可對其他結構和功能不同的多種化療藥物產生交叉耐藥性的現象,即對不同結構、功能的多種化療藥物產生抵抗。MDR性的產生原因眾多,因此逆轉或繞過MDR的腫瘤治療策略也不同[2]。由于其設計上具有良好的靶向性、生物相容性,納米制劑能夠同時負載多種化療藥物和生理活性成分,實現多角度、多方面殺傷或誘導腫瘤細胞死亡,因此納米制劑在耐藥腫瘤治療中具有廣闊的應用前景[3]。本文簡要介紹了MDR產生的幾種主要產生機制以及相關納米制劑的研究進展,以期為逆轉腫瘤多藥耐藥性治療提供思路和方法。
1 多藥耐藥性的形成機制
1.1 藥物外排增加 藥物外排增加減少化療藥物在腫瘤細胞中積累量的重要機制,其中ATP結合盒(ATP-binding cassette,ABC)蛋白家族導致的化療藥物外排是誘發耐藥性產生的重要原因[4]。目前已證實多藥耐藥蛋白1(multidrug resistance protein 1,MDR1)、多藥耐藥相關蛋白1(multidrug resistance-associated protein 1,MRP1)和乳腺癌耐藥蛋白(breast cancer resistance protein,BCRP)與許多腫瘤產生多藥耐藥性有關[5]。例如,MDR1在癌變的結腸、肝臟、腎臟細胞中表達量顯著多于正常組織,阿霉素治療可誘導肺癌細胞MDR1表達量大幅增加[5]。
1.2 腫瘤抑制基因失活,產生凋亡等死亡抵抗 腫瘤抑制基因(tumor suppressor genes,TSGs)喪失功能是導致腫瘤細胞產生多藥耐藥性的另一重要原因。TSGs是一個廣義概念,任何通過影響基因組完整性、細胞周期和細胞增殖等方式發揮腫瘤抑制作用都可定義為TSGs,其功能喪失將導致腫瘤細胞凋亡、自噬、增殖、細胞周期、DNA損傷修復和信號轉導等生理活動不再受細胞正常調控,同時可能伴隨化療藥物的脫靶、藥效降低,導致多藥耐藥性產生[6]。例如,伊馬替尼的靶點BCR-ABL在細胞質中過表達,激活PI3K/AKT通路,阻止細胞凋亡,導致耐藥性[7];厄洛替尼、吉非替尼治療導致Shisa3基因下調,通過FGFR/AKT/mTOR軸增加腫瘤細胞的干細胞特性,產生治療抵抗[8]。
1.3 上皮間充質轉化等微環境因素 腫瘤發生上皮間充質轉化(epithelial-mesenchymal transition,EMT)[9]、腫瘤相關成纖維細胞(cancer-associated fibroblasts,CAFs)[10]、腫瘤相關巨噬細胞(tumor-associated macrophages,TAMs)[11]等微環境因素能夠導致多種信號通路發生一系列變化,這些通路變化直接或間接發揮腫瘤“保護”作用,使得化療效果變差,是MDR的重要產生機制。如腫瘤招募人類間充質干細胞(MSCs)或成纖維細胞并誘導、激活其轉化為CAFs[12],CAFs中持續激活NF-κB使得TGF-β、整聯蛋白β1、IL-6等信號分子過量表達,這些上游信號可通過調節TGF-β/SNAIL、EGFR/ERK、STAT3/NF-κB等通路誘導EMT,上調耐藥相關蛋白表達,促進腫瘤細胞遷移和侵襲等[11];腫瘤細胞自身可分泌IL-4、IL-10、IL-13和乳酸等細胞因子,誘導TAMs極化為支持腫瘤生長、轉移、產生耐藥性的M2表型[13]。
2 多藥耐藥性腫瘤的治療策略
由于納米制劑通常可搭載多種化療藥物或生物活性組分,可同時克服MDR產生的多重機制,因此,納米制劑將在多藥耐藥性腫瘤治療中發揮重要的作用。
2.1 抑制藥物外排 利用納米制劑遞送外排抑制劑分子或者小干擾RNA(siRNA),是抑制外排的常用手段。目前P-gp抑制劑發展至第四代,其作用機制主要有作為P-gp底物競爭性抑制外排、抑制P-gp與ATP的結合等。代表性抑制劑有維拉帕米、他莫昔芬、奎尼丁、Elacridar等,但是親和力低和特異性差限制了P-gp抑制劑的臨床使用[14]。納米制劑不僅可以實現P-gp抑制劑的靶向遞送,還可以其對減輕對正常組織的影響。如Zhong等[15]報道了一種葉酸介導的納米紅細胞系統,并用于共同遞送化療藥物紫杉醇與Tarquidar。由于P-gp主導的外排作用,與僅搭載紫杉醇納米制劑相比,共載納米制劑在MCF-7/Taxol中的蓄積量提高了2倍,說明Tarquidar有效增加了紫杉醇在MCF-7/Taxol細胞中的積累量;此外,納米制劑可顯著增加細胞活性氧(ROS)和丙二醛(MDA)水平、降低超氧化物歧化酶(SOD)和過氧化氫酶(CAT)活性,誘導細胞氧化應激而死亡。TPGS是一種合成的維生素E衍生物,其可以通過抑制ATP酶活性、改變線粒體膜電位影響細胞呼吸等途徑,非競爭性抑制藥物外排[16]。如Assanhou等[17]構建了一種TPGS與透明質酸雙功能化納米脂質體,用以共同遞送PTX與化療增敏劑氯尼達明(lonidamine)。TPGS與氯尼達明協同作用于線粒體細胞呼吸,大幅抑制消耗ATP的藥物外排;體內、體外實驗中,與普通脂質體、分別單獨載藥脂質體相比,雙載制劑可以明顯抑制MCF-7/ADR細胞的生長。第四代抑制劑的開發仍以構效關系為指導,尋找結構適宜的化合物,目前主要處于化合物篩選與藥理研究階段[14]。
使用小干擾RNA(siRNA)等手段沉默相關基因表達是更為直接的抑制外排手段。例如ABC蛋白的表達受多種上游通路調節(見表1),抑制上游通路的納米制劑將會實現“一石多鳥”的治療效果。如Gli是Hedgehog信號通路中的一種轉錄因子,已被證明是維持膠質母細胞瘤干細胞干性的重要因子,在腫瘤侵襲與耐藥發生過程中扮演著重要角色[21]。Melamed等[22]開發了一種搭載Gli siRNA的聚乙烯亞胺-核酸納米粒(PEI-SNAs),抑制Hedgehog/Gli通路,使膠質母細胞瘤ABCG1蛋白表達下降30%、提高細胞對低劑量治療劑Temozolomide的敏感性;此外,沉默Gli基因逆轉腫瘤細胞的干細胞性,削弱其侵襲性與自我更新能力,削弱化療抵抗。
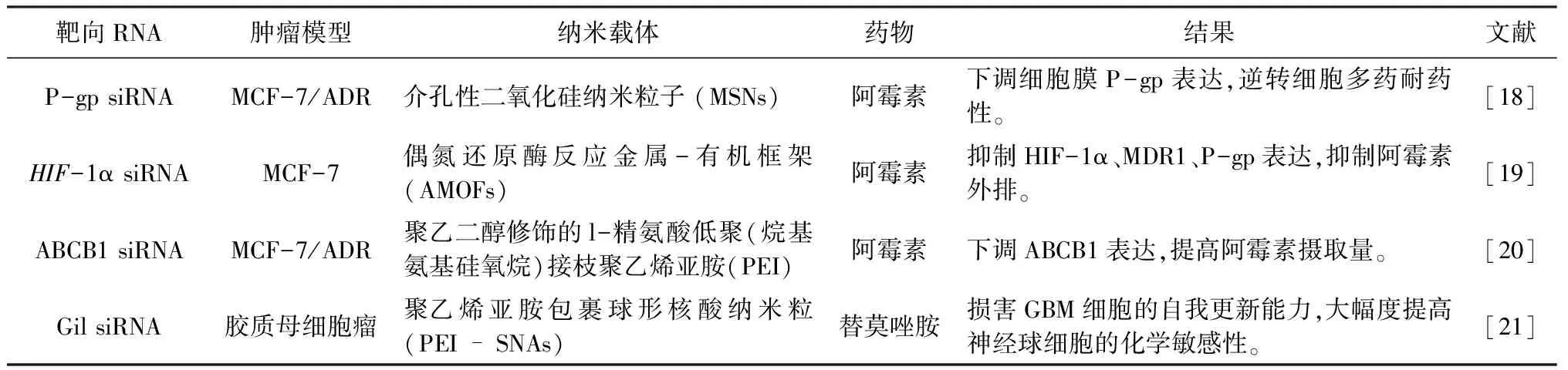
表1 以外排基因為靶點、聯用化療藥物的納米制劑示例
然而,當腫瘤細胞中的化療藥物靶點發生變異,抑制外排、增加化療藥物蓄積或將不再起效[5],這可能是外排抑制劑與化療藥物聯用臨床試驗效果有限的重要原因;此外,腫瘤細胞會對部分外排抑制劑產生耐藥性[23]。因此,在給予化療藥物及外排抑制劑的同時、聯合運用其他組分誘導細胞凋亡等細胞其他途徑死亡進程發生,將是未來克服腫瘤多藥耐藥研究中更有希望的策略。
2.2 誘導細胞其他途徑死亡
2.2.1 誘導細胞凋亡 凋亡可分為外源性凋亡和內源性凋亡。外源性凋亡主要由腫瘤壞死因子相關的凋亡誘導配體(TRAIL)誘導發生,為腫瘤壞死因子(tumor necrosis factor,TNF)超家族的一員,其可以在多種癌細胞中觸發凋亡。Miao等[24]設計了一種包載sTRAIL質粒的魚精蛋白-DNA復合物,并將腫瘤相關成纖維細胞作為 “工廠”分泌sTRAIL,誘導人胰腺癌細胞凋亡。然而,TRAIL療法的外源性本質使得腫瘤細胞可能對其再次產生耐藥性,而納米制劑可同時負載TRAIL與化療藥物,起到“1+1>2“的效果。Xu等[25]報道了一種共同遞送pTRAIL和莫能菌素(Monesin)的納米載藥系統,低分子量PEI與包載莫能菌素的環糊精通過二硫鍵交聯,而腫瘤歸巢肽RGD修飾的聚-γ-谷氨酸(γ-PGA)能夠與腫瘤相關γ-谷氨酰轉肽酶(GGT)特異性結合、實現靶向遞送與更高的遞送效率。與僅搭載pTRAIL納米粒相比,共載納米粒在HCT8/ADR細胞中的IC50值降低60%以上,細胞凋亡比例增加近一倍。
內源性凋亡受PI3K/AKT、Bcl-2/BAX等多個通路調控(見表2)。如Bcl-2/BAX通路通過上調線粒體中細胞色素C的釋放、激活caspase家族等途徑誘導凋亡[30],而PI3K/AKT是一個典型的凋亡抑制通路,可直接下調BAD等Bcl-2家族蛋白表達[31]。Diao等[32]報道了一種以姜黃素作為化療藥物增敏劑的給藥系統,將姜黃素分子化學修飾于透明質酸分子的羧基上,再將其作為載體材料包載阿霉素或紫杉醇,體外實驗結果顯示姜黃素與兩種化療藥物在A549/ADR耐藥細胞具有顯著的協同作用,給予載藥納米粒后,A549/ADR耐藥細胞P-gp表達降低、Bcl-2與Bax表達顯著上調。再如mTOR(雷帕霉素靶蛋白)是PI3K/AKT通路的下游靶點,其下游分子包含HIF-α、FOXO3等調節細胞生長與物質代謝的關鍵分子,Dai等[33]設計了一種雷帕霉素膠束與阿霉素脂質體協同給藥的治療策略,雷帕霉素顯著上調阿霉素的細胞毒性,并通過抑制mTOR、下調與MD-MB-231細胞耐藥關系緊密的HIF-α表達,從而克服三陰性乳腺癌對阿霉素的耐藥性。
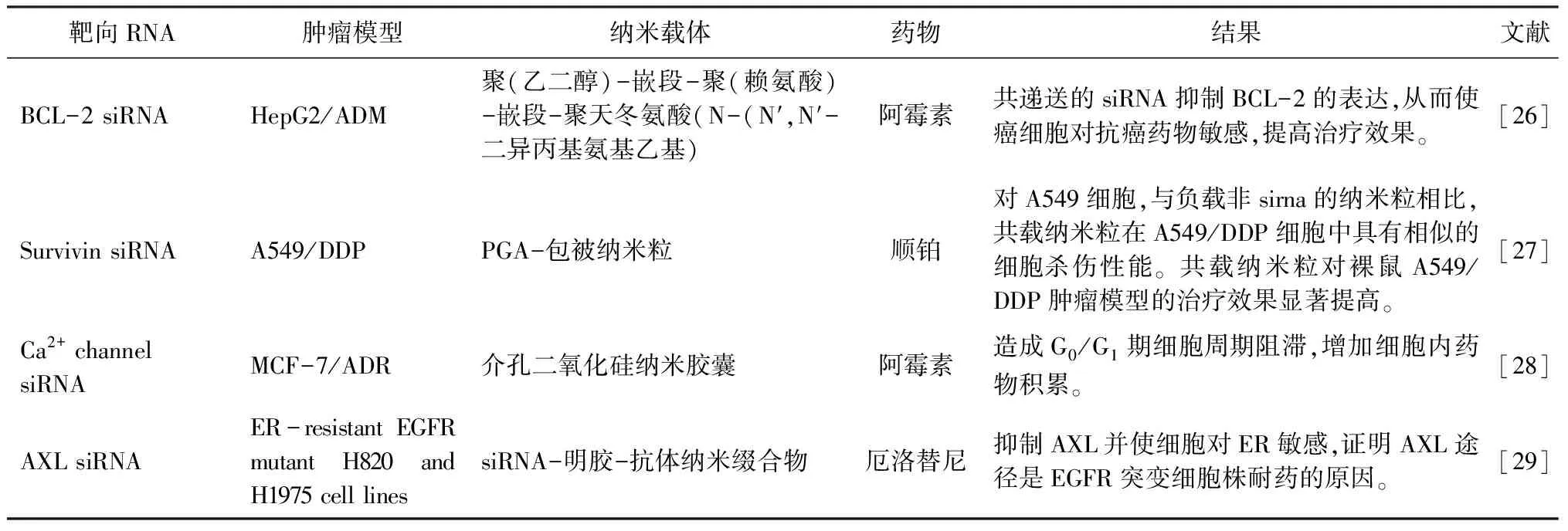
表2 以細胞程序性死亡相關基因為靶點、聯用化療藥物的納米制劑示例
2.2.2 誘導免疫原性細胞死亡 阿霉素、米托蒽醌、順鉑等化療藥物[34]通過上調細胞內ROS、引發內質網應激等方式,使腫瘤細胞釋放或暴露損傷相關分子模式(damage-associated molecular patterns,DAMPs),使得腫瘤細胞具有免疫原性,激活免疫系統對腫瘤細胞的攻擊。傳統化療藥物誘導ICD的能力有限,使用納米制劑手段搭載進行聯合治療往往能發揮更強的協同作用。Xu等[35]報道了一種基于多響應性肽的前藥納米平臺,同時搭載順鉑、Adjudin(ADD)和FRP-1激動劑WKYMVm。在MCF-7/ADR細胞中,順鉑和ADD聯合治療使順鉑的IC50值降低95%,有效逆轉了MCF-7/ADR細胞對順鉑的耐藥性;與分別給藥相比,MCF-7/ADR細胞鈣網蛋白暴露水平增加近1倍,證明誘導ICD可逆轉多藥耐藥。淫羊藿苷(icaritin)可誘導腫瘤細胞自噬、凋亡等多種細胞死亡途徑,Yu等[36]發現與阿霉素摩爾比為1∶2時可發揮最佳的誘導ICD作用,并由此構建了一種PLGA-PEG-AEAA納米顆粒實現二者的協同遞送,在人和小鼠肝細胞癌模型中,納米粒顯示出顯著的誘導ICD效果,幾乎完全抑制腫瘤生長,證實誘導ICD療法對化療和傳統免疫治療耐受的腫瘤有較強的治療能力。
2.2.3 ROS與誘導細胞死亡 細胞內活性氧(reactive oxygen species,ROS)包括羥基(·OH)、過氧基(·OOR)、硝基自由基(NO·)以及非自由基過氧化氫(H2O2)、脂質氫過氧化物(R-O-OH)等,是誘導細胞凋亡的重要因素[37]。光動力療法(photodynamic therapy,PDT)已被證明是有效的抗腫瘤策略,近年來已有多種光敏劑藥物獲批臨床使用或正進行臨床試驗[38]。光敏劑在被特定波長的光照射后,與氧氣發生反應產生單線態氧等ROS,引發細胞凋亡、自噬及壞死以殺傷腫瘤。然而,PDT對氧氣的大量消耗使后續治療中ROS產生量降低、直接影響療效,同時缺氧引發HIF-1α基因過表達,使P-gp等外排蛋白表達增加[39]。對ROS的妥善利用是克服腫瘤多藥耐藥的重要思路。
ROS水平增高引發細胞的抗氧化防御(antioxidant defense),而谷胱甘肽過氧化物酶4(GPX4)是調節該機制的關鍵因素[40]。鐵死亡是近年來廣受關注的程序性細胞死亡模式,表現為鐵依賴性的、過量的脂質氫過氧化物積累和細胞膜上不飽和脂肪酸(如花生四烯酸)的消耗[41]。作為與凋亡截然不同的細胞死亡模式,誘導細胞鐵死亡可以與凋亡途徑協同作用,克服復雜誘因產生的耐藥。Fu等[42]設計了一種搭載高鐵酸鹽和阿霉素的中空介孔二氧化硅納米粒子,通過超聲激活以實現腫瘤部位靶向釋放。其中高鐵酸鹽可與水反應生成氧氣,緩解細胞缺氧;高鐵酸鹽可與阿霉素通過再氧合(reoxygenation)產生過氧化氫反應生成毒性更強的超氧自由基,被還原產生的二價鐵繼續參與芬頓反應生成羥基自由基、消耗不飽和脂肪酸造成鐵死亡;此外,高鐵酸鹽可將還原型谷胱甘肽(GSH)氧化為谷胱甘肽(GSSH),納米粒通過改善缺氧,抑制HIF-1α基因表達,繼而下調MDR1基因和P-gp蛋白在缺氧性Saos-2骨肉瘤細胞中的表達,逆轉缺氧導致的腫瘤多藥耐藥。Gao等[43]開發了一種搭載GPX4抑制劑的納米膠束,膠束材料中的花生四烯酸可被腫瘤細胞中的ROS氧化為脂質氫過氧化物,同時具有作為響應性降解材料和誘導鐵死亡的功能;RSL3作為GPX4抑制劑,促進細胞的鐵死亡并逆轉抗氧化防御介導的對阿霉素的耐藥性。NCI-ADR/Res細胞對阿霉素極度耐受,IC50值高于100 μmol·L-1,而納米膠束IC50值僅為1 μmol·L-1左右,證明誘導鐵死亡是殺傷耐藥細胞的有效途徑。
2.3 針對CAFs/EMT的逆轉多藥耐藥的療法 上皮-間充質轉化(epithelial to mesenchymal transition,EMT)指上皮細胞通過一定程序轉化為具有間質表型細胞的過程[44]。在腫瘤發生過程中,EMT表現為整合素(integrins)、鈣粘蛋白(cadherins)表達降低以降低細胞黏著,促進轉移[45];此外,整合素αvβ1、β3能夠促進TGF-β、VEGF等因子分泌,利于腫瘤細胞增殖、存活[46]。CAFs除了自身過表達的IL-6、IL-8多種信號分子可誘導耐藥相關蛋白上調、促進腫瘤細胞遷移和侵襲外,其與EMT現象通過多個通路構成復雜緊密的關系,如IL-6/JAK2/STAT3、TGF-β/SNAIL等[11,47],因此干擾、切斷腫瘤微環境與腫瘤細胞的串擾是克服CAF/EMT誘發耐藥性的重要手段(見表3)。Jin等[48]報道了一種辛伐他汀(simvastatin)與紫杉醇共載的納米脂質體,脂質體外層以腫瘤相關豆莢蛋白酶響應的細胞穿透肽修飾,使得脂質體可在腫瘤微環境中特異性降解并同時作用于腫瘤細胞與腫瘤相關巨噬細胞。辛伐他汀可破壞脂閥、降低膽固醇水平以抑制整合素-β3/FAK通路,逆轉A549T細胞對紫杉醇的耐藥性;同時,辛伐他汀促進巨噬細胞M2表型復極化為M1表型,使得TNF-α增加、TGF-β降低,實現重塑腫瘤微環境、逆轉EMT的目的;與游離藥物相比,脂質體制劑顯示出了顯著的治療效果優勢。Wnt 16是一種損傷響應(damage response program,DRP)分子,由CAFs通過激活NF-κB或化療藥物攝取等途徑啟動DRP而分泌,可導致腫瘤對多種藥物產生耐藥性。Hu等[51]制備了一種磷酸槲皮素(quercetin)脂質顆粒,作為天然抗纖維化物質,槲皮素下調Wnt16分泌達50%,在富含基質膀胱癌模型中顯示出了與順鉑納米制劑的協同作用、逆轉Wnt16介導的順鉑耐藥性,同時實現重塑腫瘤微環境、增加納米粒對腫瘤組織滲透,提高治療效果。

表3 針對CAFs/EMT的逆轉多藥耐藥的納米制劑示例
3 克服多藥耐藥納米藥物的載體設計
與傳統制劑相比,納米藥物可以在注射入體內后、在體循環中保持穩定,保護藥物不受血漿蛋白結合等方式破壞、降低被代謝的風險,實現治療組分在腫瘤部位的高效蓄積并發揮功能。因此,設計具有獨特功能性和體循環穩定性的納米載體,是設計克服多藥耐藥納米藥物的關鍵指導策略。
3.1 共載小分子的納米藥物的設計 脂質體具有制備簡單、脂材選擇多樣等特點,對于水溶性、脂溶性藥物都能夠具有高效的包載能力,是設計納米藥物的常見選擇。Xu等[52]制備了一種共載吖黃嘌呤(ACF)與化療藥物阿霉素的脂質體。所制備的脂質體能夠實現吖黃嘌呤與阿霉素同時高效包載,包封率均高達90%以上,粒徑為140 nm左右、分布集中,可在37 ℃保持穩定12 h以上;作為缺氧誘導因子HIF-1抑制劑,吖黃嘌呤能夠抑制阿霉素誘導的HIF-1α上調,降低下游的VEGF、GLUT-1及MMP-9表達,遏制CT26細胞對阿霉素的耐藥性。
與脂質體相比,自組裝膠束材料能夠以多樣手段修飾其他組分以實現還原響應、pH響應、光熱敏感等功能,具有逆轉MDR能力的分子可直接修飾于膠束材料上、實現理想的靶向性。Xing等[53]構建了一種光敏納米系統。MDR逆轉劑TPGS具有兩親性,其疏水端包裹IR780形成膠束并作為光源,繼而負載還原敏感性光敏劑納米粒與DOX,構成多功能納米系統。納米系統受到腫瘤細胞的弱酸性環境、強還原性環境和外部近紅外照射后逐級分解,TPGS抑制外排、提高光敏劑與DOX療效,對MCF-7/ADR細胞具有強效殺傷作用。
3.2 共載核酸+化療藥物的納米藥物的設計 與使用小分子抑制劑相比,使用siRNA等核酸分子直接沉默多藥耐藥相關基因作用更加直接,且更不易產生治療抵抗。然而,核酸分子往往親水性較強,而化療藥物多為脂溶性,難以共載;核酸分子在細胞內容易被核酸酶分解、難以進入細胞核或線粒體發揮作用[54]。因此,設計遞送核酸的納米藥物,應實現核酸分子的高效包載,同時具有適宜程度的胞內穩定性,保證化療藥物等組分釋放的同時,核酸被靶向遞送至細胞核或細胞器。Wu等[55]合成了一種葉酸修飾的PEG-b-(PCL-g-PEI)-b-pcl三嵌段共聚物,其具有核-殼結構能夠保護所搭載的P-gp siRNA免受腎臟清除與RNA酶降解,并通過靜電作用實現P-gp siRNA和阿霉素的共同包載,在體內外實驗中均顯示出對MCF7/ADR細胞多藥耐藥性的顯著遏制。Wang等[56]制備了一種金納米粒,其表面修飾寡聚DNA序列、能夠與MDR1 mRNA互補;納米粒進一步被RNA酶修飾,實現對MDR1 mRNA的靶向降解,實現多藥耐藥逆轉。寡聚DNA序列保護RNA酶不受蛋白酶降解,同時規避了siRNA的不穩定性、實現對MDR1 mRNA的特異性沉默。金屬有機框架(metal-organic framework,MOF)納米粒是搭載核酸藥物納米藥物的熱門選擇。其中,有機框架材料具有一定的共軛結構與孔隙以搭載金屬納米粒;金屬納米粒作為配體,常帶有正電荷、能夠以靜電作用穩定荷載并保護siRNA分子,并發揮磁性響應[57]、誘導細胞死亡等功能[58]。Chen等[18]構建了一種硒/釕納米粒子修飾的金屬有機框架MIL-101(Fe),用以搭載多種siRNA。框架可以保護siRNA免受核酸酶降解、實現溶酶體逃逸,下調P-gp、VEGF表達;硒、釕表現出一定的細胞毒性與抗腫瘤轉移活性。框架通過微管動力學破壞,誘導細胞周期停滯及細胞凋亡,作用機制與紫杉醇類似,而對耐紫杉醇的MCF-7/T細胞具有較強抑制作用,證明MOF納米粒是理想的克服多藥耐藥策略。
4 總結與展望
由于其結構設計多樣,能夠實現靶向性遞送、體內長循環保護有效成分不被降解、協同遞送、靈活載藥量等功能,納米制劑在耐藥腫瘤治療中具有廣闊的應用前景,相較于游離藥物更易實現腫瘤部位蓄積,納米制劑實現重塑腫瘤微環境、破除腫瘤的“保護屏障”的效果,逆轉腫瘤多藥耐藥性。但穩定性差、生產工藝復雜、批間差異大等缺陷限制了納米制劑的大范圍臨床應用;外排抑制劑、凋亡誘導劑等組分對正常細胞影響較大,更易產生未知的毒副作用,對納米藥物的靶向性要求更高;腫瘤細胞由于靶點變異、代謝增強等原因,可能對逆轉MDR組分再次產生治療抵抗,限制了逆轉MDR納米藥物的臨床療效;此外,協同遞送各個組分的分子機制與代謝命運研究是納米制劑應用的重大挑戰[1]。
猜你喜歡
保健醫苑(2022年5期)2022-06-10 07:46:38
現代臨床醫學(2022年3期)2022-06-06 07:59:40
昆明醫科大學學報(2022年1期)2022-02-28 07:43:40
天津醫科大學學報(2021年3期)2021-07-21 09:04:02
科學大眾(2020年12期)2020-08-13 03:22:22
云南醫藥(2019年3期)2019-07-25 07:25:10
現代檢驗醫學雜志(2016年1期)2016-11-12 13:19:40
國外醫藥(抗生素分冊)(2016年6期)2016-07-10 11:34:45
中國衛生標準管理(2015年14期)2016-01-15 02:58:37
中國當代醫藥(2015年17期)2015-03-01 02:03:58
