小團簇吸附活化原子氧的密度泛函研究
2022-12-03 08:55:00朱裔榮陳文昊頡雨佳
湖南工業大學學報 2022年5期
關鍵詞:結構
劉 葉,袁 佩,朱裔榮,陳文昊,頡雨佳
(1.湖南工業大學 材料與先進制造學院,湖南 株洲 412007;2.湘潭大學 化工學院,湖南 湘潭 411105)
1 研究背景
計算化學具有節約研究成本、縮短研究時間、預測新材料等特性,為開發新材料提供了理論依據。3d 過渡金屬的物理和化學性質,決定了它在合金、鋰電池、表面吸附和化學催化等方面的重要應用。黃釗文等[1]采用CASTEP(Cambridge Sequential Total Energy Package)程序,以Si-Co 合金作為鋰離子電池負極材料,進行了分子設計模擬研究。結果表明,其導電性能會直接影響鋰離子的輸運和大倍率充放電性能,Si2Co具有較低的體積膨脹系數(71.151 5 %),以及較低嵌鋰的形成能(0.457 1 eV),故具有較佳的電化學綜合性能。Nong J.等[2]采用Dmol3程序,研究了Li-O2電池中氮摻雜碳基材料的結構與電子輸運性能間的關系,發現未摻雜的單層石墨烯由于其電荷分布良好而沒有活性位點吸附O2,因而吡啶-N 比石墨-N 對O2具有更高的催化活性。L.M.Jiménez-Díaz 等[3]采用Quantum Espresso 程序,對分子氧在Au12M(M 為 Cu、Ag、Ir)團簇上的吸附作用和解離作用進行了研究。其結果表明,金納米團簇中添加過渡金屬原子可以增強對CO 氧化反應的催化活性。Li T.T.等[4]采用VASP(Vienna Ab-initio Simulation Package)軟 件,分 析 了12 種Cu13-mNim(m=0,1,13)團簇的結構,結果表明,對稱性最高的Cu12Ni團簇比Cu13和Ni13團簇更易吸附O2和CO 分子,因而有利于CO 氧化。Lei X.等[5]采用Gaussian 09 軟件,系統研究了分子氧和原子氧在Ag(100)表面氧化CO 的過程,結果表明,分子氧和原子氧對CO 氧化的活化能分別為28.8,8.78 kJ/mol,說明對CO 氧化都有很強的反應活性,但分子氧和原子氧在氧化過程中的氧化機理各不相同。Hao F.等[6]采用Dmol3程序,計算了環己基過氧化氫(cyclohexyl hydroperoxide,CHHP)的O—O 鍵長和變形能,結果表明鈷負載的氮摻雜載體更容易使CHHP 解離,且計算結果與實驗觀測結果較吻合,實驗得知催化劑鈷(Co-N-rGO)催化環己烷的轉化率為8.85%,選擇性為85.73%。上述研究結果均表明,快速、環保的模擬計算可高效指導新材料的設計與合成[7]。
團簇可被作為介于原子與宏觀物質之間的橋梁,是研究宏觀催化體系的理想模型,不同尺寸的團簇可表現出不同于體材料的效果[8-9]。團簇的活性與結構、制備方法及載體等因素密切相關,識別團簇的真正活性位點變得非常困難。材料的研發成本和能耗較高、耗時較長等問題長期存在,為彌補材料合成中的問題,采用建模模擬計算材料的最優結構,指導改進合成現有的材料成分或設計新的催化劑顯得尤為重要。設計高活性和高選擇性的新型催化劑材料的關鍵,在于充分認識反應活性位點的結構與反應活性的關系。原子氧(O)和金屬鈷(Co)之間的相互作用是金屬表面氧化、表面腐蝕及催化反應等過程中至關重要的影響因素,但已有文獻中,對帶電鈷團簇吸附活化原子氧的研究較少。因此,本文擬采用密度泛函理論系統研究(n=1~5;q=0,+,-)團簇和團簇吸附原子氧后所得(n=1~5;q=0,+,-)團簇的幾何結構、穩定性能和電子性質,考察電荷對原子氧吸附活化行為的影響,以期利用模擬計算結果為吸附、防腐蝕和催化應用(CO 氧化、烷烴氧化)等實驗研究提供一定的理論指導。
2 計算方法
采用Dmol3程序包進行密度泛函理論計算,若無特殊說明,所有計算過程結果均由下列參數設置得到:自旋限制、廣義梯度近似(general gradient approximation,GGA)的Perdew-Burke-Ernzerhof(PBE)泛函,其核處理方式采用全電子,基組為雙數極化(double-numerial basis,DNP)。
幾何優化中,所有結構都保持弛豫,對稱性不限制,精度選擇fine,位移和能量分別設為0.005 ? 和1×10-5Ha(1 Ha=27.211 6 eV),直至收斂。在此條件下計算O2鍵長,為1.227 ?,與實驗值1.207 ?[7]間的誤差較小。這一結果說明,所選用的方法和參數合理可靠。
通過文獻分析[10-11],設計團簇結構,并進行優化,以獲得具最低吸附能的結構。基于優化所得團簇不等價的可能頂位(T,top)、橋位(B,bridge)、空位(H,hole)位置上(見圖 1),初始垂直距離大于3 ? 的位置處預置原子氧,再在相同條件下進行結構優化,并對所有優化后的結構(和)進行頻率計算,無虛頻[12],以確保結構是最低能量的穩定結構,之后計算其相應性質。
原子氧(O)吸附在團簇上的吸附能定義如下:
E(O)為原子氧的能量。
原子氧吸附在團簇上的吸附能Ea為負值,表示吸附放熱,其絕對值越大,吸附能力越強。
3 結果與討論
由圖3 可以明顯觀察到,各個團簇的結合能均隨著團簇尺寸的增加而增大,且隨著n增大變化趨勢逐漸趨于平緩,表明團簇的穩定性隨團簇尺寸的增加逐漸增強。對比圖3 中的曲線可以得知,陽離子型團簇()的平均結合能遠遠大于中性型()和陰離子型()團簇的結合能,這表明團簇失去一個電子后,可以顯著增強該團簇的穩定性;而和團簇的結合能變化差異不大,但相較而言的結合能要稍低于的,這表明團簇得到一個電子后其穩定性略有降低。
最高占據分子軌道(highest occupied molecular orbital,HOMO)和最低未占據分子軌道(lowest unoccupied molecular orbital,LUMO)與化學反應活性有關。HOMO 是能量最高的軌道,可以提供電子以形成一個鍵,而LUMO 是能量最低的空軌道,該軌道上可以得到更多的電子。帶隙能Egap為LUMO軌道與HOMO 軌道間的能量差值,是一個動力學穩定性指標,較大的帶隙能意味著較高的動力學穩定性和較低的化學反應活性[8]。簡而言之,帶隙能可以反映電子被激發的難易程度以及被激發時所需能量的大小,其值越大,表示該分子越難以激發,分子活性越差。電荷對團簇最高占據分子軌道(HOMO)和最低未占據分子軌道(LUMO)的能量影響順序由大到小依次為負電荷、中性、正電荷。團簇的帶隙能如圖 4 所示。
由圖8 所示各團簇的吸附能可以得知,除原子氧吸附后的Co2O T 團簇和Co2O B 團簇外,其余團簇的吸附能均隨著原子數目的增加而呈現出規律性的變化,表現為,吸附能為負值表明吸附放熱,其絕對值越大,吸附能力越強。由此說明,團簇對原子氧的吸附強度由大到小依次為。其中,所有結構中B 團簇對原子氧的吸附能力最強,其吸附能為-8.375 eV。對比中性團簇,負電荷的引入,有利于增強團簇對原子氧的吸附作用;而正電荷的引入,更容易削弱團簇對原子氧的吸附作用。
團簇的反應活性、穩定性,與電子結構和團簇中原子的數量和排列密切相關。因此,采用Mulliken電荷對原子之間的電荷轉移進行分析。團簇中原子氧電荷的變化情況如圖 10 所示,帶隙能變化情況如圖11 所示。
因原子氧的電負性(3.44)遠大于鈷原子的電負性(1.88),故原子氧對電子吸引的能力較強。鈷團簇結構吸附原子氧之后,團簇中O 得電子而帶負電,金屬團簇失電子而帶正電,電荷轉移進一步表明團簇活化了原子氧[13]。
由圖10 可看出,電荷對團簇吸附原子氧的作用呈現出規律性變化,金屬團簇帶負電時,O 原子與其作用最強,中性次之,金屬團簇正電荷時作用最弱,氧原子電荷轉移強度大小順序為。相同的n數目條件下,帶正電的團簇和中性團簇中,O 的得電子能力由小到大依次為頂位、橋位、空位;帶負電的團簇,O 得電子的能力由大到小依次為頂位、橋位、空位。出現這一結果的原因,與團簇中相應吸附位置原子氧的配位數有關,O 原子與初始的團簇頂位、橋位和空位的配位數分別為1,2,3,經幾何優化之后,部分結構(如Co2O T 團簇和Co3O B 團簇等)的配位數發生變化,與電荷協同作用的結果造成O 原子在團簇不同吸附位置處具有不同的得電子能力。
表1 O、和 B 的HOMO 和LUMO 軌道Table 1 HOMO and LUMO orbital of O, andO B
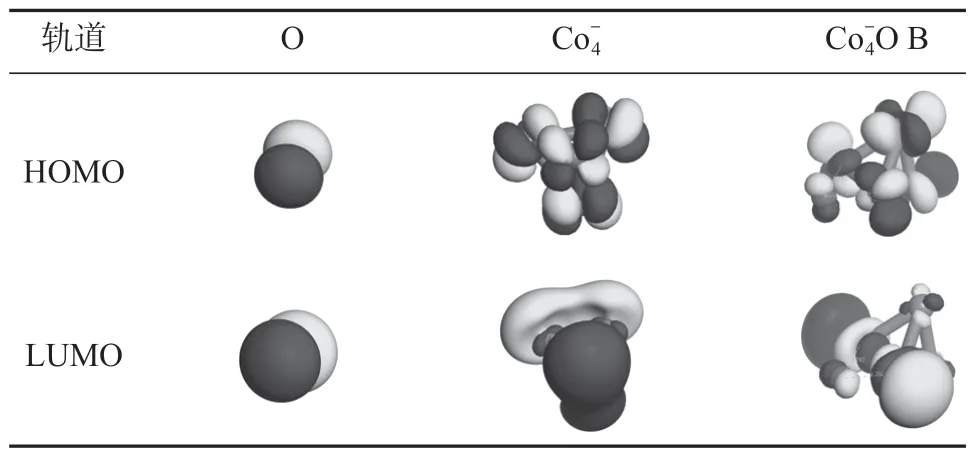
表1 O、和 B 的HOMO 和LUMO 軌道Table 1 HOMO and LUMO orbital of O, andO B
由表1 中的軌道情況可以看出,因O 是強電子接受體,其HOMO 軌道和LUMO 軌道都為p 型軌道;團簇為d 型軌道,其HOMO 軌道主要構成成鍵軌道,而LUMO 軌道主要由反鍵軌道構成。由B 團簇的HOMO 軌道和LUMO 軌道,可以明顯觀察到O 的2p 軌道與Co 的3d 軌道間的相互作用,這一結果進一步說明原子氧在團簇上的吸附主要是化學吸附。
4 結論
2)優化前的原子氧在初始位置相同的情況下,不同電荷的同種結構經過幾何優化,原子氧會發生遷移并且被吸附到最穩定的吸附位點上。從吸附能的計算結果中可以看出,電荷的引入顯著改變了團簇對原子氧的吸附強度。負電荷型團簇的吸附強度要比中性型團簇的強,而正電荷型團簇的吸附強度要比中性型團簇的弱,所有結構中,對原子氧的吸附能力最強的是帶負電橋位型的B 團簇,其吸附能為-8.375 eV。
3)電荷對團簇吸附原子氧的作用呈現規律性變化,金屬團簇帶負電時,O 與其作用最強,中性團簇次之,正電荷團簇最弱。
5)當團簇帶負電時,其Co—O 的鍵長要比中性團簇的長;而當團簇帶正電荷時,其Co—O 的鍵長要比中性團簇的短。所有吸附結構中,吸附后的團簇中原子氧都獲得電子而帶負電,其中負電荷型團簇得電子最多,中性型團簇次之,正電荷型團簇最少。
本研究為原子氧活化階段提供了相應的依據,結果有望為吸附、防腐蝕和催化應用(CO 氧化、烷烴氧化)等實驗研究和工業生產提供理論指導,但涉及氣敏、防腐蝕和氧化等體系,尚需進一步具體到理論研究和實驗相互驗證。
猜你喜歡
小獼猴智力畫刊(2023年4期)2023-04-23 08:49:58
哲學評論(2021年2期)2021-08-22 01:53:34
中華詩詞(2019年7期)2019-11-25 01:43:04
模具制造(2019年3期)2019-06-06 02:10:54
中學生數理化·高一版(2018年1期)2018-02-10 05:20:03
影視與戲劇評論(2016年0期)2016-11-23 05:26:01
七彩語文·寫字與書法(2016年7期)2016-07-28 21:40:22
七彩語文·寫字與書法(2016年6期)2016-07-15 19:36:34
人間(2015年21期)2015-03-11 15:23:21
現代企業(2015年9期)2015-02-28 18:56:50
