松果菊苷固體脂質納米粒的制備及其在體腸吸收特性、體內藥動學研究
2022-12-03 06:15:22決利利李曉婷王柯靜周珊珊劉艷菊
中成藥 2022年8期
決利利, 梁 婧, 李曉婷, 王柯靜, 周珊珊, 劉艷菊*
(1.鄭州工業應用技術學院醫學院,河南 鄭州 451150;2.無錫市農產品質量監測中心,江蘇 無錫 214400)
松果菊苷是肉蓯蓉、松果菊等中藥的主要有效成分之一[1-2],具有抗腫瘤、降血脂、抗氧化、肝保護、神經保護、抗炎等活性,可用于多種系統疾病的治療[1,3-5],但其溶解度僅為197.07 μg/mL,屬于極微溶解藥物,脂溶性差[6],而且體內容易代謝[4,7],穩定性不理想,導致口服吸收生物利用度低(0.83%)[5]。目前,鮮有關于松果菊苷新制劑技術的報道[6-7]。
固體脂質納米粒是采用一種或幾種脂質作為難溶性藥物載體制成的實體骨架型納米給藥系統[8],可促進藥物體外釋放及腸道吸收,增加口服吸收生物利用度[9-11]。因此,本實驗將松果菊苷制成固體脂質納米粒,采用在體腸灌流方法考察該制劑在大鼠不同腸段中的吸收差異,并研究其體內藥動學,以期為相關新制劑開發提供參考。
1 材料
1.1 儀器 安捷倫1200型高效液相色譜儀(美國安捷倫公司);MSE125P型分析天平(德國賽多利斯公司);JC-2LZF型旋轉蒸發儀(青島精誠儀器儀表有限公司);SZCL-2型磁力攪拌器(鄭州生化儀器有限公司);BT100-1J/BZ15型蠕動泵(重慶科耐普蠕動泵有限公司);M-sizer 3000型粒度分析儀(英國馬爾文儀器有限公司);JP-040PLUS型超聲波儀(深圳市寶安區沙井潔盟德力生電器商行);MS-3型漩渦混合器(海門市其林貝爾儀器制造有限公司);HNDK200-2型氮氣吹掃儀(上海琪摩電子科技有限公司)。
1.2 試劑與藥物 松果菊苷(批號111670-201904,純度98.3%)、綠原酸(批號110753-201716,純度98.5%)對照品(中國食品藥品檢定研究院);松果菊苷原料藥(批號190628,純度95%,武漢卡諾斯科技有限公司)。K-R試液(批號20201025,北京百奧萊博科技有限公司);甘露醇(批號191123,四川博利恒藥業有限公司);大豆磷脂(批號20190919,大連華農豆業科技發展有限公司);單硬脂酸甘油酯(批號20181208,上海吉至生化科技有限公司)。
1.3 動物 清潔級SD大鼠,雌雄不限,體質量(220±20)g,購自河南省動物實驗中心,實驗動物生產許可證號SCXK(豫)2016-0001。
2 方法與結果
2.1 松果菊苷含量測定
2.1.1 色譜條件 Venusil XBP-C18色譜柱(150 mm×4.6 mm,5 μm);Phenomenex C18預柱(4 mm×2 mm,5 μm);流動相乙腈-0.4%磷酸(60∶40);體積流量1.0 mL/min;柱溫30 ℃;檢測波長333 nm。
2.1.2 供試品溶液制備 取1 mL固體脂質納米粒混懸液至50 mL量瓶中,加入10 mL甲醇超聲提取5 min,靜置20 min,流動相定容至刻度,搖勻,過0.45 μm微孔濾膜,即得。
2.1.3 線性關系考察 取20.20 mg對照品至100 mL量瓶中(減重法稱量),乙腈定容至刻度,得202 μg/mL貯備液,密封后置于冰箱中保存,取適量,流動相稀釋至20.2、10.1、2.02、1.01、0.505、0.050 5 μg/mL,即得對照品溶液,在“2.1.1”項色譜條件下進樣測定。以對照品質量濃度為橫坐標(X),峰面積為縱坐標(Y)進行回歸,得方程為Y=17.215 3X-0.419 4(r=0.999 9),在0.050 5~20.2 μg/mL范圍內線性關系良好。
2.1.4 方法學考察 取“2.1.2”項下供試品溶液,于0、3、6、9、12、24 h在“2.1.1”項色譜條件下進樣測定,測得松果菊苷含量RSD為1.06%,表明溶液在24 h內穩定性良好。按“2.1.2”項下方法平行制備6份供試品溶液,在“2.1.1”項色譜條件下進樣測定,測得松果菊苷含量RSD為1.67%,表明該方法重復性良好。取0.050 5、2.02、20.2 μg/mL對照品溶液,在“2.1.1”項色譜條件下進樣測定,測得松果菊苷含量RSD分別為1.14%、0.38%、0.49%,表明儀器精密度良好。取9份固體脂質納米粒混懸液,每份0.5 mL,置于9個50 mL量瓶中,分為3組,分別加入202 μg/mL貯備液0.5、1、1.5 mL,按“2.1.2”項下方法制備供試品溶液,在“2.1.1”項色譜條件下進樣測定,測得松果菊苷平均加樣回收率分別為101.72%、99.46%、100.85%,RSD分別為0.62%、1.17%、1.92%。
2.2 固體脂質納米粒制備 采用冷均質法制備[8,12]。取0.8 g單硬酯酸甘油酯、0.4 g大豆磷脂,65 ℃水浴加熱熔融,加入50 mg松果菊苷,磁力攪拌溶解得熔融液,迅速置于-65 ℃超低溫冰箱中12 h得固體物質,適當粉碎后置于120 mL 1%泊洛沙姆188溶液(4 ℃)中,膠體磨中形成粗分散體系,在壓力60 MPa條件下循環均質8次,過0.45 μm微孔濾膜,加蒸餾水至120 mL,即得。再以5%甘露醇為凍干保護劑,加到固體脂質納米粒混懸液中,搖勻,在-30 ℃下預凍2 d,迅速置于凍干機中,抽真空后冷凍干燥1 d,得到凍干粉。
2.3 包封率、載藥量測定 取固體脂質納米粒混懸液1 mL,按“2.1.2”項下方法制備供試品溶液,測定松果菊苷總量W0;取1 mL固體脂質納米粒混懸液至離心管中,4 ℃、12 500 r/min離心30 min,取上清液,測定游離松果菊苷量W1;將沉淀物在-30 ℃下預凍2 d,迅速置于凍干機中,抽真空后冷凍干燥1 d,測定松果菊苷和脂質總量W,計算包封率、載藥量,公式分別為包封率=[(W0-W1)/W0]×100%、載藥量=[(W0-W1)/W]×100%。結果,3批固體脂質納米粒混懸液平均包封率為(83.34±1.41)%,載藥量為(3.19±0.23)%。
2.4 粒徑、Zeta電位測定 取固體脂質納米粒混懸液0.1 mL,蒸餾水稀釋40倍后測定其粒徑、Zeta電位。結果,固體脂質納米粒混懸液平均粒徑為(192.07±9.14)nm,PDI為0.107±0.013,而其凍干粉復溶后兩者分別為(237.77±9.14)nm、0.186±0.024,見圖1;兩者Zeta電位分別為(-22.64±2.13)、(-18.71±2.24)mV,見圖2。
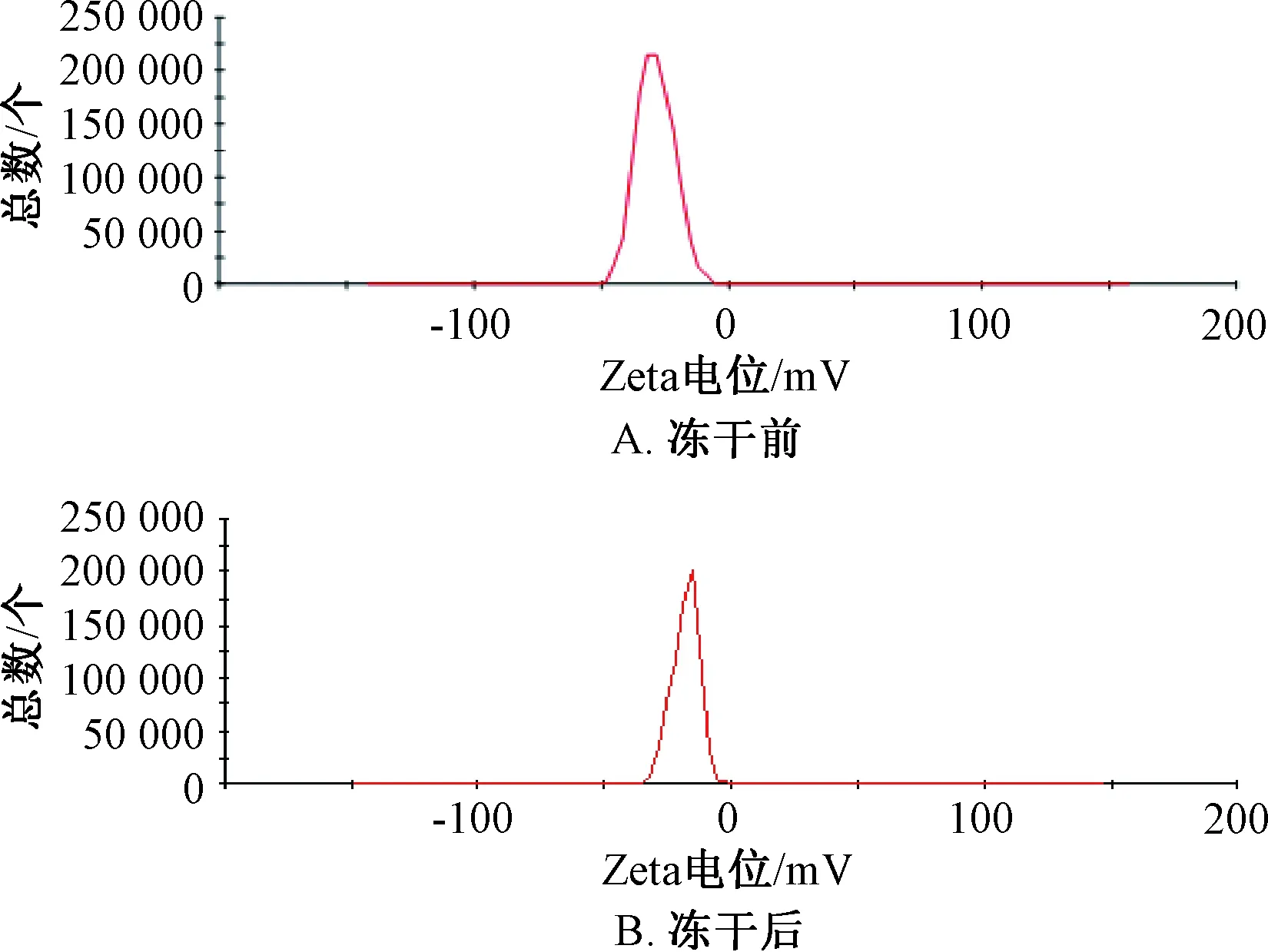
圖2 松果菊苷固體脂質納米粒Zeta電位
2.5 XRPD掃描 取松果菊苷、空白輔料、物理混合物(松果菊苷與空白輔料比例同固體脂質納米粒凍干粉)、固體脂質納米粒凍干粉適量,設定掃描范圍為3°~45°,速度為6°/min,結果見圖3。由此可知,松果菊苷在10°~30°范圍內出現多處晶型峰;物理混合物在11.9°、16.3°、16.8°、25.8°等處仍可見其晶型峰;固體脂質納米粒凍干粉僅見空白輔料的晶型峰,松果菊苷晶型峰均消失,表明該成分變為無定型物質。
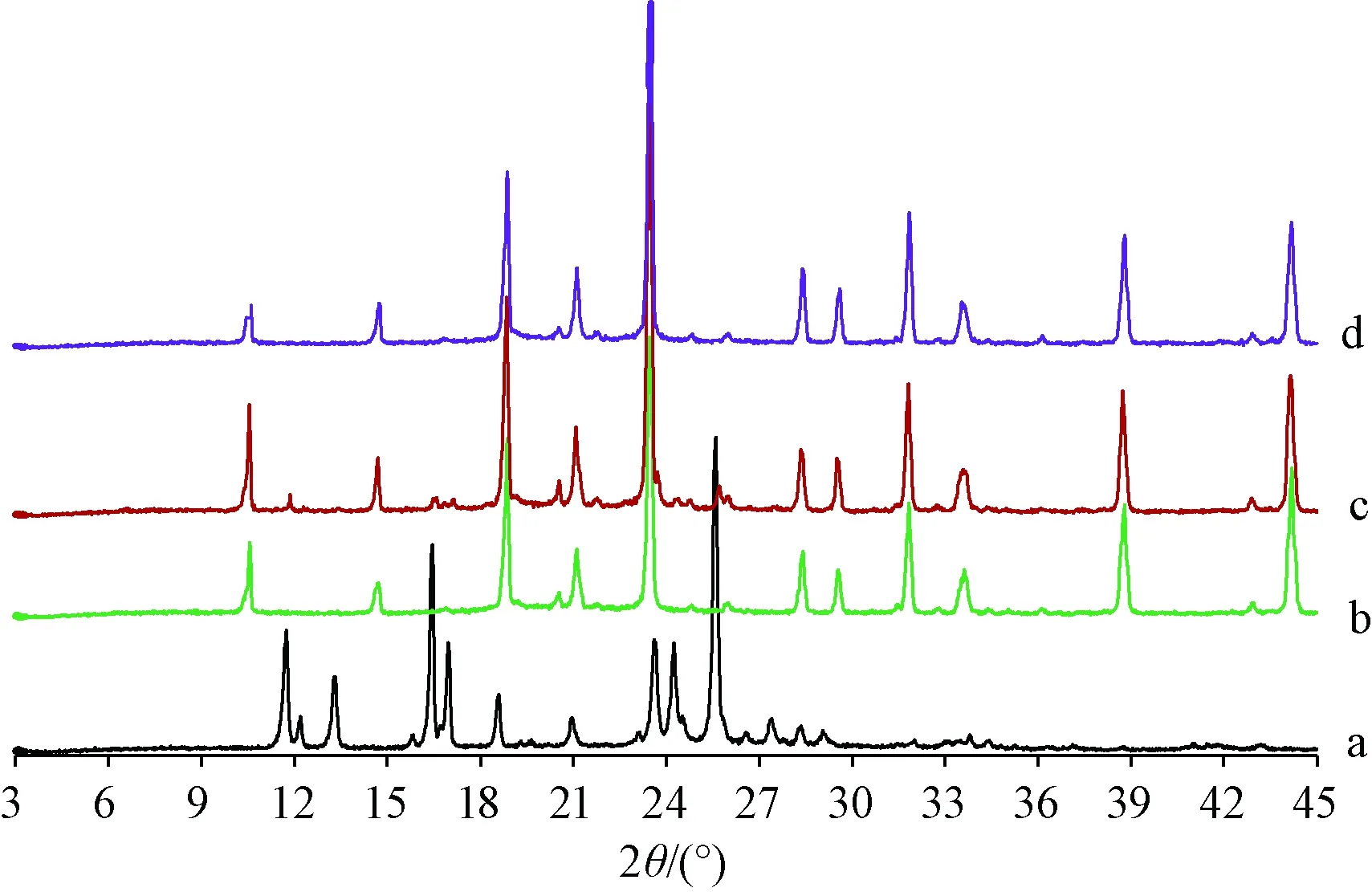
注:a~d分別為松果菊苷、空白輔料、物理混合物、固體脂質納米粒凍干粉。
2.6 油水分配系數測定 參考文獻[13-14]報道,取過量松果菊苷、物理混合物、固體脂質納米粒凍干粉適量,加入正辛醇飽和的水相,超聲處理30 min,固定于振蕩器上,37 ℃、100 r/min振蕩1 d,取上層混懸液,過0.22 μm水膜,測定在水中的溶解度C1,同法測定在蒸餾水飽和正辛醇中的溶解度C2,計算油水分配系數P,公式為P=(C1-C2)/C2,并計算lgP。結果,松果菊苷、物理混合物、固體脂質納米粒凍干粉lgP分別為0.106、0.139、0.807,表明固體脂質納米粒可增加松果菊苷油水分配系數,有助于該成分透膜吸收。
2.7 體外釋藥研究 取適量松果菊苷及其固體脂質納米粒凍干粉(含30 mg松果菊苷),加入3 mL空白釋放介質得混懸液,轉移至透析袋中(截留分子量12~14 kDa),一端用細尼龍繩扎緊,另一端固定于攪拌槳上,釋藥介質為1 000 mL蒸餾水,設定轉速、溫度分別為75 r/min、37 ℃,于0、0.5、0.75、1、1.5、2、3、4、6、8、12、18、24、36 h各取樣3 mL,補加空白釋放介質,過0.45 μm微孔濾膜,測定松果菊苷含量,計算累積釋放度,結果見圖4。由此可知,在8 h,原料藥釋放度達94.48%,而固體脂質納米粒僅為65.04%,屬于快速釋藥期;在8~36 h,固體脂質納米粒累積釋放度呈緩慢增加趨勢,36 h內達84.06%。
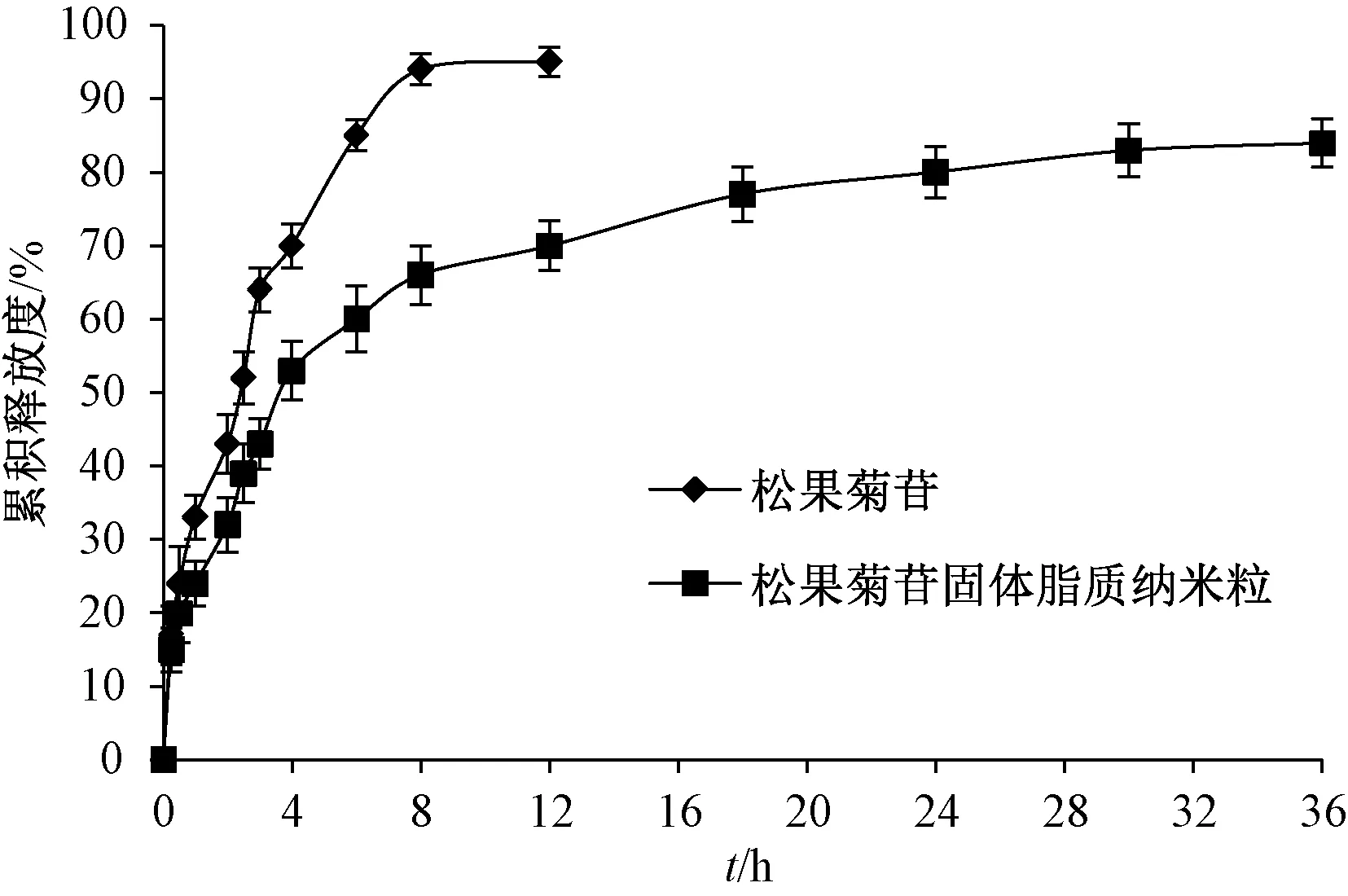
圖4 松果菊苷體外釋藥曲線(n=6)
2.8 在體腸吸收特性研究
2.8.1 腸灌流溶液制備 取K-R試液進行在體腸灌流操作,收集流出液,作為空白腸灌流液。取松果菊苷及其固體脂質納米粒凍干粉適量,空白腸灌流液配制15 mL(以松果菊苷計,質量濃度為40 μg/mL),即得。
2.8.2 含藥灌流液穩定性 取“2.8.1”項下含藥灌流液,置于37 ℃恒溫水浴中,于0、15、30、45、60、90、120 min各取樣1 mL,過0.45 μm微孔濾膜,在“2.1.1”項色譜條件下進樣測定。結果,原料藥、固體脂質納米粒灌流液峰面積RSD分別為0.71%、1.14%,表明含藥灌流液在K-R試液中穩定性良好。
2.8.3 腸壁對灌流液中藥物的物理吸附 剪取腸段,K-R試液沖洗3次,置于松果菊苷及其固體脂質納米粒灌流液中孵育2 h,K-R試液沖洗2次,分別測定孵育前和孵育2 h后松果菊苷剩余藥量。結果,原料藥、固體脂質納米粒灌流液剩余藥量分別為98.74%、101.26%,RSD分別為0.62%、0.81%,表明大鼠腸壁對灌流液中藥物無明顯的吸附作用。
2.8.4 實驗操作 取禁食12 h(可自由飲水)的大鼠12只,隨機分為松果菊苷組、松果菊苷固體脂質納米粒組,腹腔注射戊巴比妥鈉(50 mg/kg)麻醉,仰臥固定于37 ℃保溫墊上,實驗用管道均在同濃度灌流液中過夜浸泡,沿腹部中線打開腹腔,分離十二指腸、空腸、回腸、結腸,在待灌流腸段上部和下部用手術剪開口(勿剪斷),插管后扎緊,K-R試液沖洗內容物。大鼠腹部覆蓋浸有生理鹽水的紗布(適時灑生理鹽水以保濕),設置灌流液體積流量為0.2 mL/min,開啟蠕動泵,收集流出液,1 h后停止灌流,K-R試液沖洗2次,合并流出液,采用重量法對其體積進行校正。剪斷灌流結束腸段,記錄內徑(r)、長度(l),流出液6 500 r/min離心30 min,測定松果菊苷質量濃度,計算吸收速率常數Ka、表觀吸收系數Papp,公式分別為Ka=(1-CoutQout/CinQin)Q/V、Papp=-Qln(CoutQout/CinQin)/2πrl,其中Qin、Qout分別為進、出灌流液體積,Cin、Cout分別為進、出灌流液濃度,Q為灌流液體積流量,V為待考察腸段體積。
2.8.5 結果分析 表1顯示,松果菊苷在各腸段中均有一定程度的吸收,其中結腸段吸收較差;將該成分制成固體脂質納米粒后,Ka、Papp在各腸段中均升高(P<0.01),其中結腸段更明顯,推測可能存在結腸靶向。

表1 松果菊苷吸收參數
2.9 體內藥動學研究
2.9.1 血漿處理 參考文獻[2]報道,取血漿100 μL至5 mL離心管中,加入50 μL內標(綠原酸)溶液(10 μg/mL),渦旋15 s后加入1 mL甲醇沉淀蛋白,密封后渦旋3 min,8 500 r/min離心6 min,移取上層有機相至另一離心管中,置于40 ℃氮吹儀中緩慢吹干得殘渣,加入100 μL乙腈超聲處理30 s進行復溶,進樣分析。
2.9.2 分組、給藥與采血 取松果菊苷適量,加入3 mL 0.5%CMC-Na制成混懸液(6 mg/mL),同法制備固體脂質納米粒凍干粉混懸液。取禁食12 h的大鼠12只(雌雄兼具),分為2組,灌胃給予2種混懸液(50 mg/kg),于0.25、0.5、1、1.5、2、3、4、6、8、12 h采血約0.3 mL,置于涂抹肝素的離心管中,振蕩混勻,3 000 r/min離心3 min,保存于-15 ℃冰箱中。
2.9.3 線性關系考察 大鼠麻醉后心臟采血,置于涂抹肝素的離心管中,振蕩混勻,3 000 r/min離心3 min,取上清液,即為空白血漿。取松果菊苷對照品適量,空白血漿制成1 600、800、400、200、100、20 ng/mL血漿對照品溶液,按“2.9.1”項下方法處理,在“2.1.1”項色譜條件下進樣測定。以對照品質量濃度為橫坐標(X),峰面積為縱坐標(Y)進行回歸,得方程為Y=0.016 2X+0.110 7(r=0.996 3),在20~1 600 ng/mL范圍內線性關系良好。
2.9.4 方法學考察 取給藥固體脂質納米粒后1 h的血漿,于0、2、4、6、8、12 h在“2.1.1”項色譜條件下進樣測定,測得松果菊苷含量RSD為8.61%,表明血漿在12 h內穩定性良好。取20、400、1 600 ng/mL血漿對照品溶液適量,在“2.1.1”項色譜條件下進樣測定6次,測得松果菊苷含量RSD分別為9.36%、5.19%、6.04%,表明儀器精密度良好。取上述3種溶液適量,按“2.9.1”項下方法處理,在“2.1.1”項色譜條件下進樣測定,測得松果菊苷含量RSD分別為10.17%、7.56%、8.90%,表明該方法重復性良好。取上述3種溶液適量,在“2.1.1”項色譜條件下進樣測定,測得松果菊苷平均加樣回收率分別為90.14%、95.62%、92.38%,RSD分別為7.16%、4.31%、5.28%。
2.9.5 結果分析 圖5、表2顯示,與原料藥比較,固體脂質納米粒tmax延長(P<0.01),Cmax、AUC0~t、AUC0~∞升高(P<0.01),相對生物利用度提高至3.82倍。
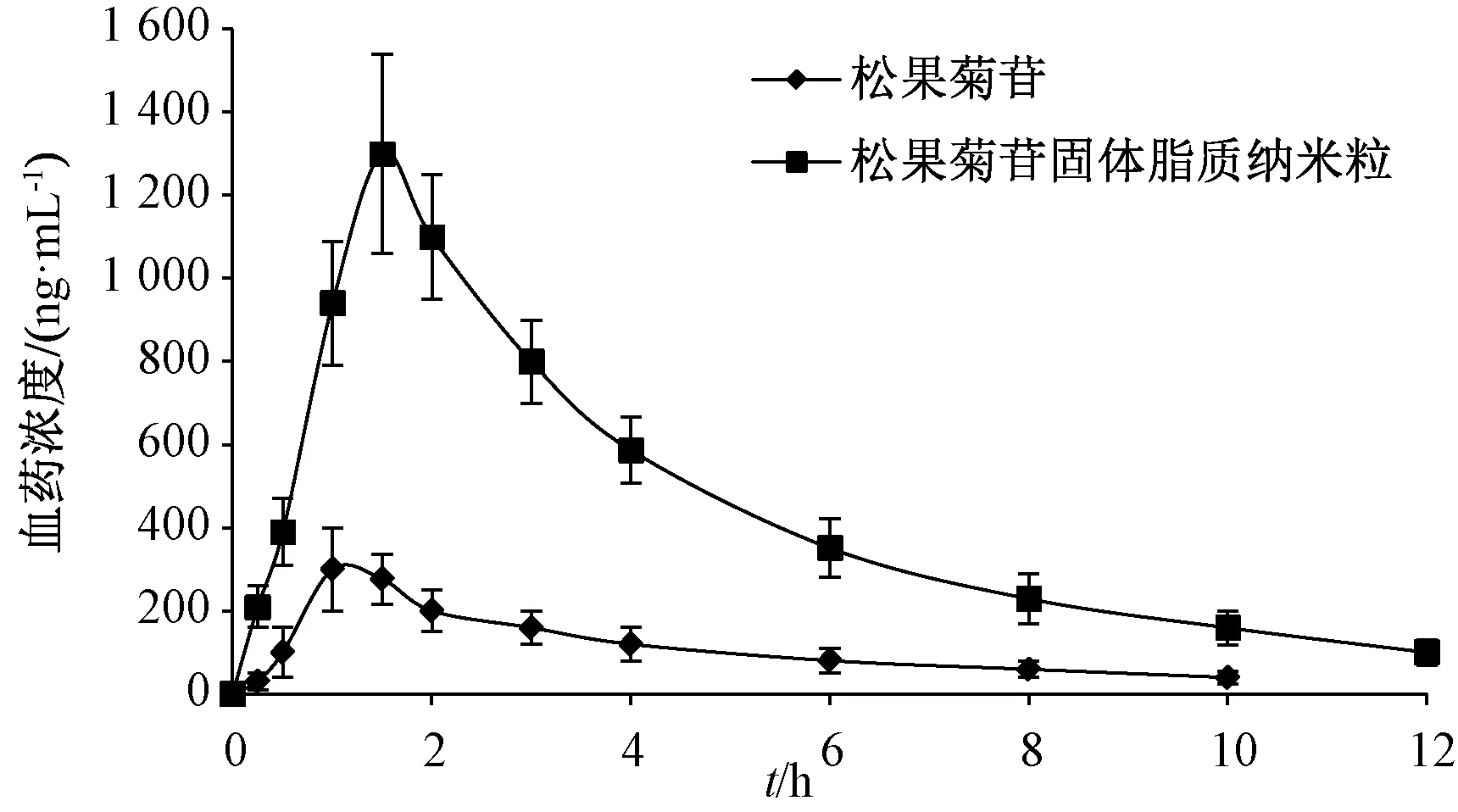
圖5 松果菊苷血藥濃度-時間曲線(n=6)

表2 松果菊苷主要藥動學參數
3 討論
陳靜等[15]采用乳化固化法制備松果菊苷固體脂質納米粒,但其包封率不足60%,可能是藥物處方中含有表面活性劑,在超聲增溶作用下使藥物進入水相,從而影響脂質載體對藥物的包裹效率。本實驗采用冷均質法制備該制劑,無需超聲提取,有助于減少藥物向水相的轉移[8],包封率達(80.24±1.53)%。
由于在體腸灌流實驗時大鼠各個腸段除了吸收藥物外,同時還吸收水分,故本實驗采用重量法對灌流體積進行校正,以保證實驗結果的準確性。結果,松果菊苷固體脂質納米粒Ka、Papp在大鼠結腸中的升高程度最大,可能是該處存在大量淋巴液[16],從而增加該成分淋巴轉運所致。
一般認為,當Papp小于3×10-6時,藥物吸收較差[17],而松果菊苷各腸段Papp均小于該數值,提示腸道吸收受限是該成分口服吸收較差的原因之一,而本實驗將其制成固體脂質納米粒,可顯著提高該參數。本實驗發現,松果菊苷在固體脂質納米粒中變為無定型物質,該形態往往比晶型藥物具有更高的生物利用度[18-19];固體脂質納米粒提高了松果菊苷油水分配系數,也有助于促進該成分吸收[14,20],從而提高其口服吸收生物利用度;與原料藥比較,固體脂質納米粒tmax延長,可能是因為脂質載體的包裹作用及胃腸道對納米藥物的吸附作用,導致部分藥物釋放延緩,從而影響tmax。
