EZH2基因敲除對局灶節段性腎小球硬化小鼠足細胞損傷和JAK2/STAT3信號通路的影響
2022-12-09 09:04:02劉衛英隋曉露張燕子陳繼紅
實用臨床醫藥雜志 2022年21期
劉衛英, 隋曉露, 張燕子, 陳繼紅
(廣東省深圳市寶安區人民醫院 腎內科, 廣東 深圳, 518000)
局灶節段性腎小球硬化(FSGS)是原發性腎小球腎炎的常見類型,最終可進展為終末期腎病[1]。FSGS病因復雜,足細胞損傷是其發病的核心病理環節[2]。研究[3]證實,表觀遺傳修飾參與足細胞損傷的過程。組蛋白甲基轉移酶EZH2可催化組蛋白H3K27發生三甲基化,使組蛋白H3第27位賴氨酸三甲基化(H3K27me3)水平上調,抑制炎性基因表達,減輕組織炎性反應,延緩腎小球疾病的進展[4]。研究[5-6]發現, JAK2/STAT3信號通路可促進炎癥因子表達,進而促進足細胞凋亡,加重腎臟疾病進展。本研究建立EZH2基因敲除FSGS小鼠模型并觀察其腎臟足細胞病理改變和JAK2、STAT3蛋白表達水平變化,探討EZH2基因敲除對FSGS小鼠腎臟足細胞損傷和JAK2/STAT3信號通路的影響,現報告如下。
1 材料與方法
1.1 實驗動物
Cas9小鼠60只, 6周齡,雌雄各半,體質量20~25 g, 購自南京模式公司。將小鼠于恒溫環境下標準化飼養1周,隨機分為EZH2敲除組和EZH2未敲除組,每組30只。本研究經本院倫理委員會審核通過。
1.2 實驗材料
重組腺病毒(AAV9-sgRNA-EZH2), 購自上海和元公司; nephrin抗體、podocin抗體,購自Proteintech公司; TUNEL試劑盒,購自Roche公司。
1.3 小鼠模型構建
① 小鼠腎臟EZH2基因敲除及未敲除對照: 使小鼠仰臥于操作臺,麻醉小鼠。左腹部剃毛,于小鼠左腹部消毒后做縱切口,暴露左腎及左腎蒂,游離腎靜脈。將注射針刺入左腎靜脈近端,EZH2敲除組小鼠注入AAV9-sgRNA-EZH2, 病毒載量2×1011/mL,EZH2未敲除組小鼠注入磷酸鹽緩沖液(PBS)。拔針后移去顯微止血夾,壓迫止血,逐層縫合,培養30 d。② FSGS小鼠模型構建: 消毒小鼠尾靜脈后給予阿霉素(25 mg/kg)尾靜脈注射。7 d后留取尿樣,監測24 h尿蛋白定量,若24 h尿蛋白定量>100 mg, 提示EZH2敲除FSGS模型或EZH2未敲除FSGS模型構建成功。收集24 h尿液、血漿后處死小鼠,摘取雙側腎臟,取適量腎臟組織觀察其病理學改變,將剩余腎臟組織于-80 ℃凍存。
1.4 觀察指標
1.4.1 生化指標: 收集小鼠24 h尿液、血漿,檢測2組小鼠24 h尿蛋白定量和血清肌酐(Cr)、尿素氮(BUN)、天門冬氨酸氨基轉移酶(AST)、丙氨酸氨基轉移酶(ALT)水平。
1.4.2 腎臟組織形態學: ① 用10%甲醛固定腎組織后脫水、包埋、切片,蘇木素-伊紅(HE)染色,于光鏡下觀察腎組織。② 去掉腎皮質,將腎組織剪成1 mm3大小組織塊,用固定液快速固定,脫水、滲透、包埋、切片后鈾鉛雙染色,于透射電鏡下觀察腎小球基底膜、足細胞。
1.4.3 足細胞nephrin和podocin表達情況: 將腎組織切片、烤片、脫蠟、熱修復后用山羊血清封閉,滴入nephrin抗體和podocin抗體,適溫孵育過夜, PBS洗滌后滴加二抗,避光適溫孵育,洗滌后4′, 6-二脒基-2-苯基吲哚(DAPI)復染細胞核,加入自發熒光淬滅劑,封片,采用激光共聚焦技術觀察。
1.4.4 TUNEL法檢測足細胞凋亡情況: 將腎組織石蠟包埋、切片、脫蠟、水洗,先后滴加蛋白酶K及破膜工作液,取TUNEL試劑覆蓋組織,血清孵育,切片置入雙氧水溶液,避光孵育15 min, 二氨基聯苯胺(DAB)顯色,蘇木素復染,脫水封片,光鏡下觀察。
1.4.5 免疫印跡(Western blot)法檢測腎臟JAK2、STAT3蛋白表達水平: 取適量腎組織,加入蛋白質裂解液裂解 30 min后,提取組織蛋白,采用Western Blot法檢測2組小鼠腎臟JAK2、STAT3蛋白表達水平。
1.5 統計學分析
2 結 果
2.1 生化指標水平
與EZH2未敲除組FSGS小鼠比較,EZH2敲除組FSGS小鼠24 h尿蛋白定量、BUN、Cr水平更高,差異有統計學意義(P<0.01); 2組ALT、AST水平比較,差異無統計學意義(P>0.05)。見表1。
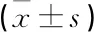
表1 2組FSGS小鼠生化指標水平比較
2.2 腎臟組織形態學觀察結果
2.2.1 光鏡: HE染色結果顯示,EZH2未敲除組病變呈局灶、分段分布,腎小球足細胞肥大,胞漿空泡樣變性,無腎小管結構病理改變;EZH2敲除組可見腎小球病變加重,節段性系膜基質明顯增多,且伴團塊狀物質沉積,毛細血管部分閉塞,可見球囊粘連,小管萎縮。見圖1。
2.2.2 電鏡:EZH2未敲除組腎小球基底膜增厚,足突排列相對整齊,足突增寬、偶有融合;EZH2敲除組腎小球足突排列紊亂,足突明顯增寬、廣泛融合甚至消失,基底膜塌陷。見圖2。
2.3 足細胞nephrin和podocin表達
nephrin和podocin陽性表達分別顯示為綠光和紅光,與EZH2未敲除組相比,EZH2敲除組nephrin、podocin表達減少,見圖3。
2.4 足細胞凋亡情況
EZH2敲除組足細胞凋亡指數為(40.94±2.13)%, 高于EZH2未敲除組的(21.23±3.30)%, 差異有統計學意義(P<0.01), 見圖4。
2.5 足細胞JAK2蛋白、STAT3蛋白表達
與EZH2未敲除組相比,EZH2敲除組JAK2、STAT3蛋白表達增加,差異有統計學意義(P<0.01)。見圖5、表2。

表2 2組FSGS小鼠足細胞JAK2、STAT3蛋白表達水平比較
3 討 論
阿霉素腎病小鼠模型的病理改變與人類FSGS病變最接近,目前己成為經典的FSGS動物模型[7]。足細胞損傷是導致蛋白尿和腎功能惡化的病理生理基礎[8]。足細胞基底膜脫落不僅能引起蛋白尿,也是發生FSGS的啟動因素[2]。nephrin是足細胞裂孔膜上重要的信號轉導分子,起著調節細胞骨架及足細胞凋亡等作用。podocin是足細胞內信號轉導必需蛋白,可促進nephrin的信號轉導。nephrin和podocin在維持足細胞裂孔膜的正常功能及足突的完整性方面起著關鍵性作用。
表觀遺傳學是指非基因序列改變所致基因表達水平的變化,主要包括DNA甲基化、組蛋白甲基化、組蛋白乙酰化等多種調控方式[9]。EZH2最早在腫瘤細胞中被發現,是多梳抑制復合體2中的催化亞基,在基因沉默和轉錄抑制中起重要作用[10]。EZH2主要通過羧基末端SET結構域催化H3K27me3, 調控靶基因轉錄水平、下游炎癥因子表達水平并激活相關信號通路[11]。研究[12]發現,EZH2低表達可引起H3K27me3水平下調,促進轉錄因子配對核基因6和硫氧還蛋白互作蛋白的表達來誘導足細胞損傷和增高氧化應激水平,提示組蛋白 H3K27me3表達水平參與足細胞損傷過程。
JAK2/STAT3信號通路主要參與機體炎癥信號轉導及細胞凋亡,代謝產物激活信號通路后,引起炎癥因子高表達,促進足細胞凋亡,在慢性腎臟病病程進展中發揮重要作用[5]。JAK2是酪氨酸蛋白激酶,通過結合受體介導相關細胞信號傳遞。STAT3是DNA結合蛋白,與酪氨酸肽段結合后形成二聚體,進入細胞核與靶基因特定位點結合,激活基因轉錄[13]。體內磷酸化STAT3表達水平上調使得EZH2活性增強、組蛋白H3K27me3表達水平上升,抑制抑癌基因的表達,促進腫瘤浸潤轉移,提示EZH2可能參與JAK2/STAT3信號通路的調控,影響細胞分化、凋亡過程。
本研究結果顯示,與EZH2未敲除組FSGS小鼠相比,EZH2敲除組FSGS小鼠的腎小球、足細胞病理改變更明顯,且尿蛋白定量、BUN、Cr水平升高,足細胞nephrin、podocin表達水平下降,腎臟JAK2、STAT3蛋白表達水平增高,足細胞凋亡指數增加,差異有統計學意義(P<0.01)。由此提示,EZH2敲除后JAK2/STAT3信號通路蛋白表達上調,加重足細胞損傷,EZH2可能參與JAK2/STAT3信號通路的激活過程。這可能是因為EZH2敲除減弱了對靶基因的抑制作用,促進JAK2酪氨酸磷酸化,同時磷酸化JAK2催化受體酪氨酸發生磷酸化修飾形成停泊位點,促進STAT3蛋白募集。磷酸化修飾后的STAT3蛋白進入細胞核內與促纖維化蛋白基因結合,造成細胞外基質增多,促進足細胞損傷[14]。研究[12]報道,抑制糖尿病腎病模型大鼠體內EZH2水平,可加重蛋白尿和足細胞病變,與本研究結果相似。體外研究[15]證實,給予足細胞EZH2抑制劑,可通過組蛋白甲基化增強JAK2/STAT3信號通路,加重足細胞損傷。
綜上所述,本研究成功構建EZH2敲除FSGS模型后發現,EZH2敲除后JAK2/STAT3信號通路相關蛋白JAK2、STAT3表達增加,足細胞nephrin、podocin表達水平顯著下降,足細胞凋亡指數顯著增加,提示EZH2可能參與JAK2/STAT3信號通路的調控并影響FSGS的進展。但EZH2基因敲除促進阿霉素誘導足細胞凋亡的潛在發病機制仍不明確,后續研究需進一步深入探討。
猜你喜歡
興趣閱讀·興趣作文與閱讀(低年級)(2025年8期)2025-08-18 00:00:00
美與時代·美術學刊(2022年3期)2022-04-27 01:18:15
鴨綠江(2021年35期)2021-04-19 12:24:18
考試與評價·高一版(2020年6期)2020-11-02 02:45:24
學苑創造·A版(2020年9期)2020-10-13 09:41:02
人大建設(2019年12期)2019-05-21 02:55:32
電子制作(2018年11期)2018-08-04 03:25:42
鑿巖機械氣動工具(2016年3期)2016-03-01 04:00:25
云南中醫學院學報(2014年3期)2014-07-31 18:57:34
七彩語文·畫刊(2012年3期)2012-04-29 00:00:00
