STING介導的炎癥在心血管疾病中的作用
2022-12-20 12:08:08聶曉妍王林濤何朝勇
藥學研究 2022年11期
關鍵詞:小鼠
聶曉妍,王林濤,何朝勇
(中國藥科大學藥學院,江蘇 南京 211198)
固有免疫反應作為機體識別、抵御病原體的第一道防線,產生大量免疫效應分子,維持機體內環境穩定。病毒及細菌等微生物侵入生物體產生的DNA、RNA、脂多糖及肽聚糖等成分被稱為病原體相關分子模式(pathogen-associated molecular patterns,PAMPs)[1];機體自身創傷、應激后釋放的DNA、高遷移率族蛋白1(high mobility group box 1,HMGB1)等物質被稱為危險相關分子模式(damage-associated molecular patterns,DAMPs)。PAMPs和DAMPs均可被模式識別受體(pattern recognition receptors,PRRs)識別,DNA感受器作為胞質中重要的PRRs,識別胞質中游離的DNA,激活固有免疫反應。
環狀鳥苷酸-腺苷酸合成酶(cyclic GMP-AMP synthase,cGAS)、DEAD-box RNA解旋酶41(DEAD-Box Helicase 41,DDX41)、干擾素誘導核蛋白16(interferon gamma-inducible protein 16,IFI16)等DNA感受器通過不同的方式激活干擾素基因刺激因子(STING)。cGAS通過合成2′3′環狀鳥苷酸-腺苷酸(2′-3′-cyclic GMP-AMP,2′3′-cGAMP)激活STING,而DDX41及IFI16直接激活STING。DNA感受器作為DNA受體及STING的上游通路元件介導了STING的活化,STING是固有免疫反應中的關鍵接頭蛋白。STING活化后通過內質網-內質網高爾基體中間體(reticulum-Golgi intermediate compartment,ERGIC)-高爾基體途徑囊泡運輸,激活下游TANK 結合激酶1(TANK-binding kinase 1,TBK1)、干擾素調節因子3(interferon regulatory factor 3,IRF3)及核轉錄因子-κB(nuclear factor-kappa B,NF-κB)等下游通路信號分子。IRF3及NF-κB可以上調干擾素(interferon,IFNs)、干擾素刺激基因(interferon stimulated genes,ISGs)及炎癥因子水平,調控心血管疾病、自體免疫疾病、腫瘤等疾病的病理進程。現就STING介導的炎癥及其在心血管疾病中的作用、STING抑制劑的發展情況進行綜述。
1 STING介導的炎癥反應
雙鏈DNA(double-strand DNA,dsDNA)通過DNA感受器激活STING。正常情況下,DNA存在于細胞核、線粒體等膜隔開的區室中,同時細胞中脫氧核糖核酸酶(deoxyribonucleases,DNases)會使逃逸DNA失活。細胞核或線粒體通透性增加、DNases失活會導致胞質dsDNA異常堆積,通過DNA感受器過度激活STING。激活的STING通過內質網- ERGIC-高爾基體途徑囊泡運輸,運輸過程中伴隨著STING的構象改變及下游通路的激活。STING在內質網形成多聚物后通過ERGIC轉運至高爾基體,在高爾基體,STING的Cys88及Cys91位點發生棕櫚酰化,從而活化TBK1并磷酸化IRF3,STING也可以直接或間接激活NF-κB。STING-TBK1-IRF3或STING-NF-κB途徑促進下游炎癥因子表達增加,介導機體內炎癥反應的發生。
1.1 STING信號通路 STING包含4個跨膜螺旋(TM1、TM2、TM3和TM4),N端細胞質配體結合域(LBD)和C端信號域(CTT)。非激活狀態下,LBD在內質網的胞質側形成二聚體,跨膜螺旋在STING二聚體中采用結構域交換結構,二聚體中的8個跨膜螺旋組織成兩層:來自兩個亞基的TM2和TM4形成中心層,外圍被TM1和TM3包圍。跨膜區域和LBD相互作用,形成完整的域交換二聚體。TM4和LBD中的首個螺旋之間的連接子(LBDα1)之間存在一個兩親性螺旋(以下稱為連接器),連接器通過連接器環連接到LBDα1形成連接器- LBDα1元件,靜息狀態二聚體中兩個連接器-LBDα1元件形成右手交叉結構。cGAMP 與STING結合后,STING發生蛋白結構重排,促使STING通過并排堆積形成寡聚體[13]。
內質網STING通過ERGIC轉運至高爾基體,在高爾基體中發生Cys88與Cys91位點棕櫚酰化進一步促進STING招募并活化TBK1及IRF3 。近期發現,高爾基體內合成的硫酸化糖胺聚糖(sGAGs)作為STING共配體介導STING在高爾基體的活化,sGAGs與STING的高爾基體內側極性氨基酸結合,像鉸鏈一樣引發STING多聚化[14]。STING在高爾基體的進一步寡聚使其具備了招募TBK1及IRF3的條件。CTT對于STING激活TBK1及IRF3是必需的,CTT中存在一個保守的共識基序 (pLxIS;p是親水性殘基,x是任何殘基),其在人STING的Ser366位點(小鼠位點在Ser365)磷酸化由TBK1介導并招募活化IRF3。另外,STING在內質網移位過程中激活NF-κB抑制因子激酶(IκB kinase,IKK),從而磷酸化IκB,使其通過泛素-蛋白酶體途徑降解,釋放出游離的NF-κB[16]。除此之外,胞質腫瘤壞死因子受體相關因子6(TNF receptor associated factor 6,TRAF6)E3泛素連接酶介導STING連接K63多泛素鏈,通過轉化生長因子β激活激酶1(TGF-β activated kinase-1,TAK1)、TAK1結合蛋白2/3(TAK1 binding protein2/3,TAB2/3)和IKK,激活NF-κB。盡管以上結果表明STING直接激活NF-κB引起炎癥反應,但也有文獻證明STING通過TBK1激活NF-κB[17]。此信號傳遞過程復雜,目前仍有待進一步解決。
此外,STING還可以在某些特殊情況下激活,如STING蛋白結構中的特殊位點發生突變、NOD樣受體家族含CARD結構蛋白3(NOD-like receptor family CARD domain containing 3,NLRC3)與DNA的結合、尼曼匹克蛋白1(Niemann-Pick disease type C1,NPC1)的缺失等。N154S、V147L、V155M等STING的突變類型可以引起STING自發活化。NLRC3與STING結合使其處于抑制狀態,DNA存在的情況下,NLRC3釋放并激活STING。NPC1是STING進入溶酶體的接頭蛋白,NPC1缺失抑制STING進入溶酶體降解,激活STING[22]。
因此,STING作為胞內重要的接頭蛋白,在多種因素刺激下均有作用,研究其下游發揮的不同作用將有利于剖析STING介導的生理功能與炎癥發生。
1.2 STING-TBK1-IRF3介導的炎癥反應 STING-TBK1-IRF3途徑的激活可能需要內質網膜蛋白胰島素誘導基因1(insulin induced gene-1,INSIG1)、SREBP裂解激活蛋白(SREBP-cleavage activating protein,SCAP)的輔助。INSIG1介導E3泛素連接酶自分泌運動因子受體(autocrine motility factor receptor,AMFR)對STING的K27泛素化,這有利于TBK1的招募[23]。STING通過SCAP 與IRF3連接,促進STING磷酸化IRF3。磷酸化的IRF3形成二聚體進入細胞核與特定基因啟動子結合促使IFNs、ISGs及炎癥因子表達上調(見圖1)。
NPC1缺失引起小鼠尼曼匹克病C型表型,表現為小腦中浦肯野細胞丟失、小膠質細胞激活,炎癥反應發生。NPC1敲除小鼠腦組織及巨噬細胞中的STING激活,促使趨化因子配體1(C-X-C motif chemokine ligand,CXCL1)、CXCL10、趨化因子2(C-C chemokine ligand 2,CCL2)、CCL5等炎癥趨化因子及干擾素誘導的三角形四肽重復蛋白1(interferon-induced protein with tetratricopeptide repeats 1,IFIT1)、IFIT2、IFIT3等ISGs的mRNA水平升高,血清中巨噬細胞炎性蛋白-1α(macrophage inflammatory protein -1α,MIP-1α)、MIP-1β、IL-13、嗜酸性粒細胞趨化因子(eotaxin)表達增加,STING或IRF3敲除后上述趨化因子、ISGs的mRNA水平及炎癥因子表達降低,小腦中浦肯野細胞丟失減少,小膠質細胞激活減少[22]。游離脂肪酸引起人正常肝臟細胞中STING-IRF3通路激活,肝細胞中IL-6、IL-1β等炎癥因子升高,這種升高可以通過敲除STING或IRF3逆轉。二氧化硅顆粒能引起肺部炎癥及肺纖維化,導致硅肺病的發生。硅肺病患者肺組織中STING-TBK1-IRF3途徑激活,CXCL10、IFNβ表達升高,此外在硅肺病小鼠模型中也觀察到了TNF-α、IL-1β表達增加。上述通過STING-IRF3通路介導升高的炎癥因子促進了機體內炎癥反應的發生,造成了機體不同程度的損傷。
1.3 STING-NF-κB介導的炎癥反應 STING在高爾基體上激活NF-κB抑制因子(inhibitor of NF-κB,IκB)激酶(IκB kinase,IKK)[16],磷酸化轉錄因子IκB,使其通過泛素-蛋白酶體途徑降解,釋放出游離的NF-κB。角質細胞核DNA損傷信號傳到胞質腫瘤壞死因子受體相關因子6(TNF receptor associated factor 6,TRAF6)激活STING。在此過程中TRAF6作為E3泛素連接酶介導STING連接K63多泛素鏈,K63多泛素鏈組裝轉化生長因子β激活激酶1(TGF-β activated kinase-1,TAK1)、TAK1結合蛋白2/3(TAK1 binding protein 2/3,TAB2/3)和IKK,激活NF-κB,上調炎癥水平。盡管以上結果表明STING直接激活NF-κB引起炎癥反應,但也有文獻證明STING通過TRAF6-TBK1軸激活NF-κB[17]。綜上,STING-NF-κB是否通過TBK1介導還有待進一步闡明。NF-κB活化后進入細胞核誘導如腫瘤壞死因子α(tumor necrosis factor-α,TNF-α)、白介素-1β(interleukin-1β,IL-1β)及白介素-6(interleukin- 6,IL-6)等炎癥因子表達上調(見圖1)。
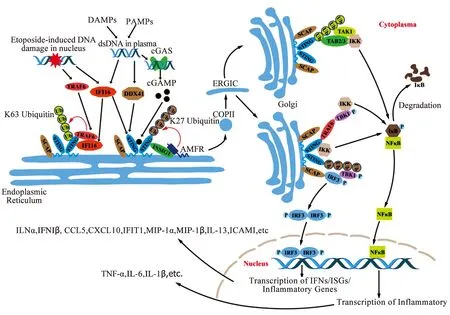
圖1 STING活化后通過TBK1-IRF3及NF-κB通路激活炎癥反應
高脂喂養誘導小鼠發生非酒精性脂肪性肝炎,該模型中肝臟枯否細胞的STING激活,NF-κB 表達水平上調,IL-6、IL-1β、TNF-α等炎癥因子水平升高,這種升高在STING敲除的小鼠中被抑制,并可被NF-κB 抑制劑BAY11-7082進一步逆轉[26]。近期在急性腎損傷與慢性腎損傷的小鼠模型中發現STING-NF-κB通路的激活。順鉑誘導小鼠發生急性腎損傷,該模型中腎小管上皮細胞線粒體損傷,線粒體DNA泄漏到胞質激活cGAS,活化STING-NF-κB途徑,上調IL-6、細胞間黏附分子-1(intercellular cell adhesion molecule-1,ICAM1)、CXCL10、粒細胞-巨噬細胞集落刺激因子(granulocyte-macrophage colony-stimulating factor,GM-CSF)等炎癥因子及趨化因子水平。腎小管細胞特異性敲除線粒體轉錄因子A(mitochondrial transcription factor A,TFAM)的小鼠作為慢性腎損傷的模型,發現該模型小鼠腎小管內的線粒體大量釋放激活STING-NF-κB通路,IL-6、IL-1β、TNF-α、CCL2等上調,導致腎臟慢性炎癥。STING-NF-κB途徑激活IL-6及TNF-α等經典炎癥因子,上調機體內的炎癥反應。
2 STING介導的炎癥促進心血管疾病病理進程
炎癥與心血管疾病的發生密切相關,近年來STING介導的炎癥反應在心血管疾病研究中取得一定進展。在心血管系統中,STING主要表達在免疫細胞、血管內皮細胞、血管平滑肌細胞(vascular smooth musle cells,VSMC)、心肌細胞及心肌成纖維細胞。細胞死亡或線粒體受損導致dsDNA釋放,激活DNA感受器,活化STING通路,介導炎癥因子轉錄上調,這可能是心血管疾病中炎癥發生的重要機制。
2.1 內皮損傷 內皮細胞作為血管炎癥中重要的驅動細胞,其在動脈粥樣硬化起始階段或慢性代謝性疾病相關的炎癥反應起著重要作用。肥胖致使外周循環中游離脂肪酸含量增加,促使血管內皮細胞線粒體DNA泄漏到胞質激活cGAS,活化STING-IRF3途徑,上調ICAM-1,導致血管內皮炎癥反應的發生。血管通透性改變是膿毒癥致死過程中重要環節,LPS引起血管內皮細胞焦亡發生,增加的胃泌素D(gasdermin D,GSDMD)活化促使線粒體DNA釋放,從而激活cGAS-STING,抑制內皮細胞增殖,最終導致血管內皮通透性改變。上述充分表明STING參與著內皮細胞炎癥反應的發生與發展。
2.2 動脈粥樣硬化 動脈粥樣硬化(atherosclerosis,AS)是一種慢性炎癥疾病,是臨床心血管事件的主因。巨噬細胞和VSMC參與了AS的炎癥發生過程。研究發現在高脂飲食喂養ApoE敲除小鼠中巨噬細胞特異性表達STING引發血管炎癥,這可能是由TDP43誘導線粒體DNA釋放介導的[31]。AS的血管斑塊中,VSMC會從收縮型轉向合成型,分泌細胞外基質(extracellular cell matrix,ECM)構成纖維帽,增加斑塊穩定性,降低心血管事件的發生。慢性腎病(chronic kidney diseases,CKD)促使VSMC過早衰老,并使其以自分泌/旁分泌的方式進行表型轉換,導致纖維帽中血管平滑肌細胞丟失,纖維帽變薄,斑塊破裂易感性增加,加速了AS的斑塊破裂進程。在上述過程中,CKD誘導的氧化應激導致VSMC線粒體損傷,線粒體通透性轉換孔(mitochondrial permeability transition pore,MPTP)開放,線粒體DNA釋放入胞漿并通過cGAS-STING通路上調Ⅰ型IFNs,激活Janus激酶(Janus kinase,JAKs)-轉錄激活因子1(signal transducer and activator of transcription 1,STAT1)信號通路,引發炎癥反應 。以上研究證明,線粒體DNA通過STING-IRF3途徑介導VSMC的炎癥反應,促進As發展。
2.3 主動脈剝離 主動脈瘤及主動脈夾層(aortic aneurysm and dissection,AAD)表現為進行性VSMC丟失及ECM碎裂、耗竭,主動脈瘤、主動脈夾層形成,最終發展為主動脈破裂。Wei等在人和小鼠的AAD樣本中發現,主動脈VSMC的dsDNA泄漏至胞質激活STING,引發細胞壞死導致dsDNA釋放入血管壁,招募活化巨噬細胞。dsDNA在巨噬細胞中活化STING-TBK1-IRF3通路,IRF3與基質金屬蛋白酶9(matrix metalloproteinase-9,MMP9)啟動子結合上調,MMP9表達增加并釋放入血管壁,破壞血管彈力板,促進AAD進程。
2.4 心肌梗死 實驗表明,過度活躍的炎癥信號會導致心肌梗死后左心室擴張增加,收縮功能障礙。心肌梗死后,骨髓及腎臟來源的單核細胞被招募至心臟,初期吞噬細胞碎片分化為促炎的M1型巨噬細胞,單核細胞對凋亡細胞的有效清除可能促使巨噬細胞偏向修復的M2型轉化。心肌梗死后心肌組織的修復對于恢復心肌功能至關重要,抑制STING通路促使心肌梗死后招募的巨噬細胞偏向修復的M2型轉化。CXCL10與巨噬細胞的M1樣極化相關,在心肌梗死模型小鼠心臟的巨噬細胞中高表達;CD163、IL-10和CCL17是巨噬細胞的M2型標記物,在cGAS敲除的心肌梗死模型小鼠的心臟中表達增加。心肌梗死的小鼠模型中STING和cGAS表達水平升高,STING-IRF3通路活化,IFNβ、CXCL10、IRF7等因子表達上調,STING敲除抑制了上述因子的表達。Lai等[39]證明了小鼠心肌缺血損傷后,心肌細胞釋放DNA及HMGB1,再灌注時二者進入循環激活炎癥反應,引起缺血再灌注損傷。在心肌缺血再灌注小鼠中使用STING抗體阻斷Ⅰ型IFNs信號通路,可顯著減小梗死面積。以上研究證明免疫細胞中cGAS監測到心肌梗死釋放的DAMPs,并通過STING-IRF3途徑介導了心肌梗死中炎癥的發生。
2.5 系統性炎癥導致的心肌損傷 STING參與介導了由二手煙、系統性紅斑狼瘡(systemic lupus erythematosus,SLE)及膿毒癥等危險因素引起的系統性炎癥,導致心肌損傷。側流煙霧在實驗中用于模擬二手煙吸入,引起小鼠心肌細胞線粒體損傷,線粒體DNA釋放入胞質,激活STING通路,誘導TNF-α和IL-1β表達水平上調,導致心臟結構、功能異常,敲除單個Beclin1等位基因加重了側流煙霧引起的炎癥反應及心功能障礙。這是因為Beclin1參與細胞自噬小體的組裝,清除細胞內大量產生的線粒體DNA,有助于抑制dsDNA誘導的STING過度磷酸化,避免炎癥反應過度發生。生理狀況下, 3′-核酸修復外切酶1 (three prime repair exonuclease 1,TREX1)清除胞質DNA,防止內源性DNA積聚,TREX1突變失活會導致系統性紅斑狼瘡(systemic lupus erythematosus,SLE)的發生[41]。SLE是一種自身免疫疾病,相較正常人SLE患者更易患心血管疾病,1/3的SLE死亡是心血管事件引起的[42]。TREX1基因敲除小鼠通過激活STING-IRF3通路產生高水平IFNs,導致心肌炎、血管炎等疾病發生,敲除cGAS可以遏制上述炎癥反應。LPS誘導小鼠膿毒性心肌病模型,表現為心功能下降、炎癥水平升高,小鼠的心臟射血分數及短軸縮短率在STING敲除后明顯改善,炎癥水平也明顯降低。膿毒性心肌病模型小鼠的發病機制是STING在LPS刺激下活化NOD樣受體熱蛋白結構域相關蛋白3(NOD-like receptor protein 3,NLRP3),NLRP3在胞質中活化半胱天冬酶-1(cysteinyl aspartate specific proteinase 1,caspase-1),切割IL-1β及IL-18,升高炎癥水平[44]。以上研究證實,上述危險因素引起的系統性炎癥中,STING介導了心肌組織炎癥反應的發生,造成心肌損傷。
2.6 化療藥物導致的心肌損傷 順鉑是廣譜化療藥物,臨床上發現順鉑可以造成心肌損傷[45]。我們的前期研究發現順鉑誘導心肌損傷的小鼠模型中TNF-α、IL-6等炎癥因子表達上調,然而,敲除Sting基因可有效抑制順鉑引起的心肌炎癥因子表達與心功能不全。在諸多臨床化療藥物治療患者中觀察到長期的化療治療經歷促使患者在多年后表現出心功能不全,STING介導的炎癥反應在化療藥物誘導心肌損傷中的作用值得獲取更多的關注。
2.7 心力衰竭 心力衰竭是心血管疾病的終末期臨床綜合征,表現為心臟收縮或舒張功能障礙,射血功能受損。擴張型和肥厚型心肌病的人類樣本中發現STING、IFNα和IFNβ水平升高。用于模擬心力衰竭的主動脈縮窄(transverse aortic constriction,TAC)小鼠模型表現為心肌肥厚、心功能不全和心肌纖維化,STING、IFNα和IFNβ的表達增加,STING敲除的TAC小鼠心功能明顯改善[46]。TAC術后3 d,小鼠心肌組織中IFNs、CXCL10、IFIT3和ISG15的表達水平顯著升高,腺相關病毒9(adeno-associated virus,AAV9)沉默cGAS可顯著減少小鼠心臟左室重構和纖維化[47]。上述研究證實了STING引起的炎癥反應促進了心力衰竭的病理進程。
2.8 STING相關的嬰兒期發病血管病變 STING相關的嬰兒期發病血管病變(STING-associated vasculopathy with onset in infancy,SAVI)是單基因突變疾病,表現為全身炎癥,嚴重的皮膚血管病變、間質性肺病及反復細菌感染。該突變促使STING在無需配體的情況下從內質網運往核周小泡中聚集活化,激活下游炎癥相關通路,上調干擾素表達,促使炎癥發生,其中STING突變體類型分為遺傳型和自發型,自發型突變較遺傳型發病早,程度也更為嚴重,如V155M。N154S、V155M是SAVI患者中常見的突變表型,相較于N153S突變類型小鼠,V154M小鼠具備更強的疾病表型,這或許解釋了SAVI患者群體中發病程度的差異性;IRF3及NF-κB通路同時參與了N153S、V154M小鼠中STING突變的下游機制[49]。臨床調查表明,除卻V147L、V147M及跨膜區中S102P等少數突變位點,大多SAVI患者的STING突變位點如N154S、V155M、V155E、R281Q、R284S、G207E等,位于STING的cGAMP結合結構域。SAVI的發生與上述STING突變引起的多聚化相關,其中V147L、N154S、V155M突變發生于STING的C148位點附近,多個STING蛋白的C148位點之間形成二硫鍵促使STING多聚化進而激活,實驗表明V147L、N154S的作用依賴于C148[51]。上述研究表明STING功能獲得性突變引起的炎癥反應促進了SAVI發生。
3 STING抑制劑
上述研究證實,STING介導的炎癥反應促進了心血管疾病的發展。因此靶向抑制STING,緩解炎癥反應過度發生,為心血管疾病的治療提供了一個新思路。現有STING抑制劑的開發主要針對STING的配體結合口袋及棕櫚酰化位點靶點進行計算機輔助設計,通過高通量篩選篩選出候選分子,并驗證候選分子對鼠源或人源STING的抑制效率。以配體結合口袋為靶點的抑制劑與STING的內源性配體cGAMP結合,抑制cGAMP對STING的激活作用,這類抑制劑主要包括SN-011、天然產物環肽Astin C、四氫異喹啉類(化合物1和化合物18)等。STING 的Cys88和Cys91位點發生棕櫚酰化對于其活化過程中形成多聚復合物、招募下游信號通路分子是必要的。僅對Cys91位點有抑制作用的抑制劑包括硝基呋喃類(C176、C178、C170和C171)、H151及丙烯酰胺類(BPK-21和BPK-25),硝基脂肪酸類(nitro-fatty acids,NO2-Fas;NO2-cLA,NO2-OA)則對兩個棕櫚酰化位點均有抑制作用。研究表明STING抑制劑緩解了小鼠As的發展,提示STING抑制劑有望成為治療心血管疾病的藥物。研究證實以上抑制劑對鼠源STING有抑制效果,目前STING抑制劑對心血管疾病的治療作用處于臨床前研究階段,其在臨床心血管疾病中的作用尚需研究驗證。
4 結語與展望
研究證實STING介導的炎癥對心血管疾病有著促進作用。心血管疾病中,免疫細胞、血管內皮細胞、VSMC及心肌細胞等發生線粒體損傷或細胞死亡,導致線粒體或細胞核DNA泄漏到胞質中。胞質中DNA感受器在dsDNA的刺激下,通過STING-TBK1-IRF3上調IFNs、ISGs及ICAM-1、MIP-1β、IL-13、IL-1β等炎癥因子水平,介導了肥胖中內皮損傷、CKD加劇的As中VSMC炎癥,加重了AAD、心肌梗死及心力衰竭;或者通過STING-NF-κB途徑增加IL-6、IL-1β、TNF-α等炎癥因子表達,可能介導了順鉑引起的心肌損傷。在心血管系統炎癥反應發生的過程中,STING起到了接頭蛋白的作用,將細胞DNA感知與炎癥反應聯系起來。因此,靶向抑制STING為治療心血管疾病提供了一個新思路。
前述STING抑制劑能否成藥取決于其對人源STING的靶向性及安全性。SN-011、C170、C171及NO2-FAs、H-151對鼠源和人源STING均有抑制作用,但H151毒性較大且對STING特異性不高,SN-011的毒性相對較低但尚未經臨床實驗證實其有效性與安全性。目前CXA-10作為安全且耐受性良好的NO2-Fas已進入Ⅱ期臨床試驗(NCT04125745、NCT04053543、NCT03449524),目的是探究其對肺動脈高壓的治療效果。除STING抑制劑外, RU.521、G150、化合物S3、A151、CU-76、阿司匹林等cGAS抑制劑及氨來占諾、化合物 II、BX795、Domainex等TBK1抑制劑也可以抑制STING通路介導的炎癥反應。其中氨來占諾是臨床上市藥物且減緩了小鼠AAD的進程,阿司匹林用于治療COVID-19患者體內STING通路活化引起的血栓性凝血功能障礙。綜上,STING通路抑制劑有望成為臨床心血管疾病的治療藥物。
猜你喜歡
興趣閱讀·興趣作文與閱讀(低年級)(2025年8期)2025-08-18 00:00:00
東方少年·布老虎畫刊(2023年8期)2023-08-01 15:45:12
科學大眾(2021年6期)2021-07-20 07:42:44
科學(2020年3期)2020-11-26 08:18:30
學苑創造·A版(2020年9期)2020-10-13 09:41:02
娃娃樂園·綜合智能(2019年3期)2019-04-03 09:17:36
中成藥(2018年2期)2018-05-09 07:19:34
小學生學習指導(低年級)(2017年10期)2017-10-10 01:00:05
湖北師范大學學報(自然科學版)(2015年2期)2016-01-10 08:41:55
云南中醫學院學報(2014年3期)2014-07-31 18:57:34
