原發性中樞神經系統非特殊類型外周T細胞淋巴瘤1例
2022-12-21 02:50:08廖智明蔡姣迪陳國群
臨床與實驗病理學雜志 2022年11期
關鍵詞:系統
符 樊,廖智明,蔡姣迪,陳國群
患者男性,39歲,2020年6月17日以突發左側面部及左上肢抽搐4次,伴舌咬傷入院。頭部CT檢查示右側額葉深部病灶,性質待定。MRI檢查示右側額葉可見大片狀T1W1稍低信號,T2W1稍高信號影,范圍約45 mm×35 mm。PET-CT檢查示該患者其他部位無明顯異常。術中B超示右額葉病灶,大小約50 mm×40 mm,顯微鏡下行保全功能區腦組織完整切除腫瘤。
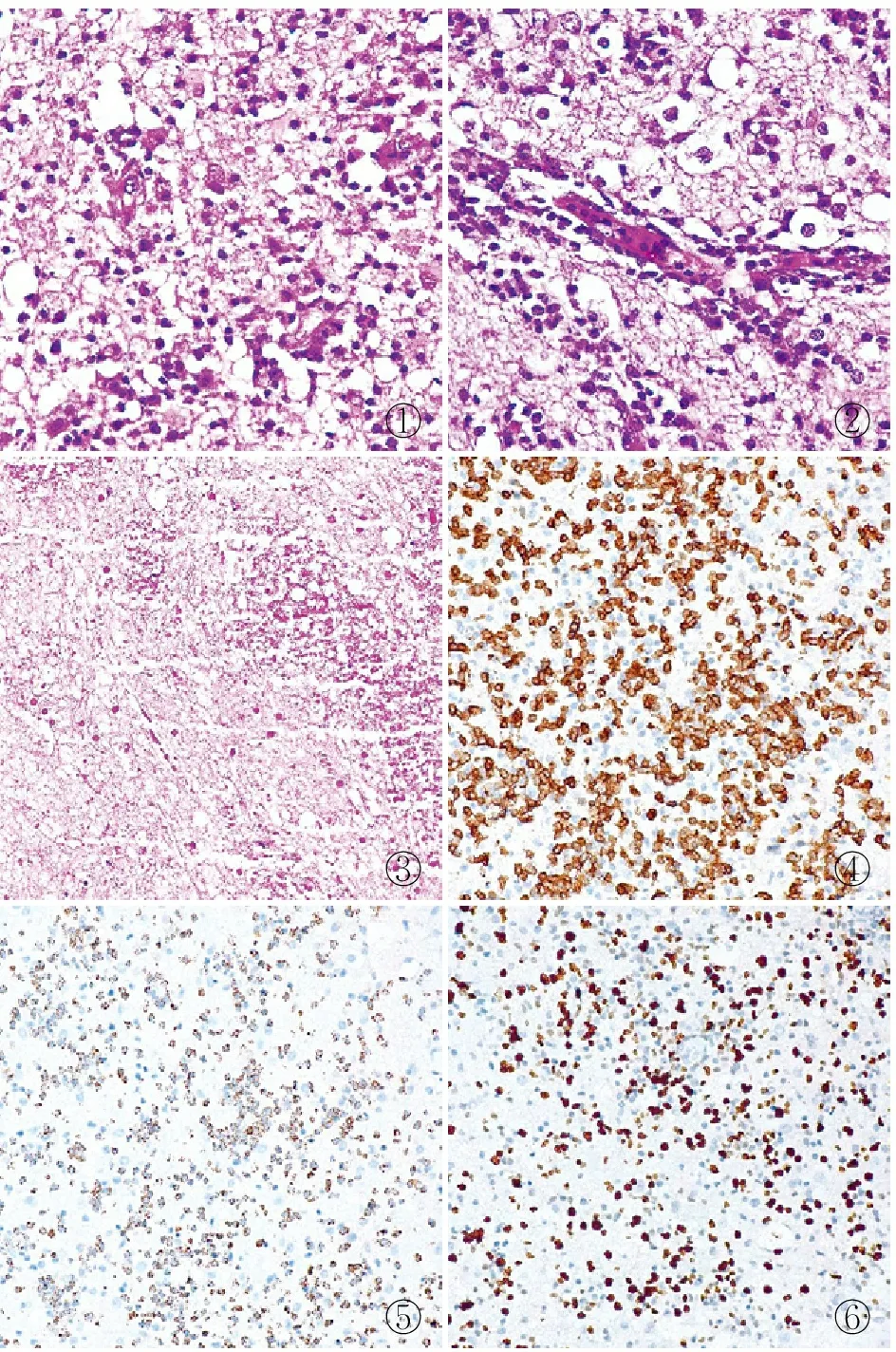
病理檢查眼觀:碎組織一堆,合計40 mm×20 mm×10 mm,呈灰白色,質軟。鏡檢:腦組織內彌漫分布大量淋巴樣腫瘤細胞,部分腫瘤細胞圍繞血管生長,形成血管周圍淋巴袖套樣外觀。高倍鏡下腫瘤細胞小~中等,異型性顯著,細胞核多形、扭曲,大部分腫瘤細胞染色質濃染,部分可見空泡狀核、核仁,核分裂象罕見,胞質較少,核質比增高,局灶可見壞死(圖1~3)。免疫表型:CD3(圖4)、CD8、TIA-1(圖5)、GrB均陽性,CD20、CD5、CD163、CD68、CD138、CD4、CD30、CD56、GFAP、SOX-10、S-100、ALK均陰性,Ki-67增殖指數約50%(圖6);EBER原位雜交陰性。TCRB基因重排檢測:TCR-β片段分析檢測有單克隆重排;TCRG基因重排檢測:TCR-γ片段分析檢測有單克隆重排。
病理診斷:原發性中樞神經系統非特殊類型外周T細胞淋巴瘤(primary central nervous system peripheral T-cell lymphomas, not otherwise specified, PCNSPTCL-NOS)。
討論原發性中樞神經系統淋巴瘤約占中樞神經系統腫瘤的2%,以彌漫大B細胞淋巴瘤最為常見,約占90%。T細胞淋巴瘤約占原發性中樞神經系統淋巴瘤的2%[1]。與西方國家相比,原發性T細胞淋巴瘤在亞洲國家,如日本、韓國發病率稍高[2]。
文獻報道59例PCNSPTCL-NOS,臨床無特殊癥狀,以頭痛多見,可有頭暈、癲癇、偏癱等。其病灶數目以單發多見,大腦多個部位均可發病,如:額葉、頂葉、基底節等。男性患者稍多于女性,比例約為1 ∶1.14,以中老年人最為常見,年齡2~89歲,中位年齡50.5歲。
①②③④⑤⑥
本例39歲男性患者診斷為PCNSPTCL-NOS,鏡下見腦組織內彌漫分布大量淋巴樣腫瘤細胞,部分腫瘤細胞圍繞血管生長。腫瘤細胞小~中等大,異型性顯著,細胞核多形、扭曲,大部分染色質濃染,可見空泡狀的核、核仁。在中樞神經系統疾病診斷中,出現血管周圍淋巴細胞套要高度懷疑惡性淋巴瘤。在PCNSPTCL-NOS中,異型的淋巴樣細胞CD3、TIA-1、GrB、CD4和(或)CD8均陽性,CD5、CD7均陰性,EBER原位雜交檢測常陰性,T細胞基因重排陽性多見。
大多數PCNSPTCL-NOS的病例來源于αβ-T細胞[3],由于PCNSPTCL-NOS發病率非常低,對其基因的研究較少。有研究表明在原發性中樞神經系統淋巴瘤中存在基因突變,有些基因的突變會影響常見的信號通路,如NFκB信號通路[4]。同時,有研究者在PCNSPTCL-NOS的腫瘤細胞中檢測到體細胞突變,其突變基因包括:DNMT3A、KRAS、JAK3、STAT3、STAT5B、GNB1和TET2,但是同一個突變的基因未在多個病例中發現,表明其在基因組學上可能具有異質性。還有學者發現,JAK/STAT通路在原發性中樞神經系統T細胞淋巴瘤中發揮重要作用。
鑒別診斷:(1)血管炎,是指血管壁破壞性的炎癥性反應,可見小淋巴細胞及其他多種炎癥細胞。(2)NK/T細胞淋巴瘤,腫瘤細胞浸潤表現為嗜血管中心性和血管破壞性,細胞核仁不明顯,免疫組化標記CD56通常陽性,EBER原位雜交陽性。(3)間變大細胞淋巴瘤,腫瘤細胞有不等量的怪異核、馬蹄鐵或腎形細胞核,免疫組化CD30通常為彌漫陽性。
PCNSPTCL-NOS常見治療方法為化療+放療,甲氨蝶呤為一線化療藥物,放療常為全腦放射治療。盡管化療和放療敏感性較高,但緩解往往是短期的,主要是因為血腦屏障限制許多藥物進入中樞神經系統。有研究表明原發性中樞神經系統T細胞淋巴瘤的中位生存期為22個月,總生存期為25個月,大多數患者在5年內復發,需要密切進行隨訪。
本例PCNSPTCL-NOS診斷需通過HE染色、免疫表型及T細胞基因重排來綜合分析。患者行手術+化療,隨訪約5個月,一般狀況較好。
猜你喜歡
工業設計(2022年8期)2022-09-09 07:43:20
軍民兩用技術與產品(2021年10期)2021-03-16 06:05:30
北京測繪(2020年12期)2020-12-29 01:33:58
裝備制造技術(2019年12期)2019-12-25 03:06:46
制造技術與機床(2019年10期)2019-10-26 02:47:06
中國洗滌用品工業(2019年4期)2019-05-11 09:27:34
鐵道通信信號(2018年5期)2018-06-28 03:06:24
家庭影院技術(2017年9期)2017-09-26 03:41:45
知識經濟·中國直銷(2017年5期)2017-06-15 20:28:19
通信電源技術(2016年6期)2016-04-20 06:21:32
