一株高產纖維素酶菌株的分離及鑒定
2023-01-09 07:16:40劉登輝趙小斌張佑紅
武漢工程大學學報 2022年6期
劉登輝,胡 閩,趙小斌,危 威,張佑紅
武漢工程大學環境生態與生物工程學院,湖北 武漢 430205
我國農作物秸稈資源豐富,是一種可再生的纖維素資源,但由于纖維素結構的難以破壞性,造成纖維素資源的利用率不高。開發利用纖維素資源,有助于解決我們當前面臨的環境污染和資源短缺問題[1]。微生物是纖維素酶的主要來源,通過微生物產生的纖維素酶高效催化降解纖維素,使得纖維素類物質糖化,然后進一步發酵生產成生物質能源酒精、有機酸等急需的食品及化工原料[2-4],且由于其生產過程經濟環保,能耗低,反應溫和等特點是當前提高纖維素利用的重要手段。
纖維素酶分為三類,即內切纖維素酶、外切纖維素酶和β-葡萄糖苷酶。酶復合體作用于纖維素的不同部分,水解機制是通過它們的協同作用將纖維素轉化為葡萄糖。目前已被研究的纖維素酶普遍存在著催化效率低,無法達到生產標準的問題。里氏木霉是研究得較為清楚的產纖維素酶菌株,但是其生長量相對較小,產酶量不能滿足工業中轉化纖維素的需求。因此,開發具有更高效纖維素酶的新菌株,具有重要的工業應用價值,對纖維素類資源的利用具有重要的實際意義[5-6]。本研究旨在從蝗蟲腸道內篩選獲得一株高產纖維素酶的菌株,用于生物乙醇工業。
1 實驗部分
1.1 化學試劑與材料
1.1.1 樣本采集蝗蟲樣本為云南省普洱市江城哈尼族彝族自治縣曼灘村采集到的蝗蟲[7-9]。
1.1.2 主要試劑羧甲基纖維素鈉(CMC-Na)、七葉苷、4-硝基苯基-D-吡喃葡糖苷等化學試劑(分析純,上海麥克林生化科技有限公司)。
1.1.3 培養基初篩培養基:CMC-Na,20 g/L;Na2HPO4·12H2O,2.5 g/L;KH2PO4,1.5 g/L;蛋 白胨,2.5 g/L;酵母浸粉,0.5 g/L;自然pH值。
剛果紅培養基:CMC-Na,1.88 g/L;Na2HPO4·12H2O,2.5 g/L;KH2PO4,0.5 g/L;MgSO4,0.3 g/L;蛋白胨,2.5 g/L;酵母浸粉,0.5 g/L;瓊脂20 g/L;剛果紅溶液,1 g/L;自然pH值。
PDA培養基:馬鈴薯浸粉,5 g/L;瓊脂粉,14 g/L;葡萄糖,20 g/L。
七葉苷培養基:蛋白胨,10 g/L;酵母浸粉,3 g/L;氯化鈉,5 g/L;七葉苷,0.1 g/L;檸檬酸鐵銨,0.2 g/L;瓊脂粉,15 g/L;自然pH值。
種子培養基:尿素,0.3 g/L;蛋白胨,0.75 g/L;酵母浸粉,0.25 g/L;(NH4)2SO4,1.4 g/L;KH2PO4,2 g/L;CaCl2,0.3 g/L;MgSO4,0.3 g/L;葡萄糖,10 g/L;吐溫80,1 mL/L和1mL/L的微量元素,自然pH值,其中,所述微量元素由如下組分組成:5 g/L的FeSO4·7H2O;1.6 g/L的MnSO4·4H2O;1.4 g/L的ZnSO4·7H2O和20 g/L的CoCl2·6H2O。
發酵培養基:CMC-Na,10 g/L;酵母浸粉,5 g/L;蛋 白 胨,10 g/L;MgSO4,0.5 g/L;NaCl,5 g/L;KH2PO4,1 g/L;吐溫80,1 mL/L和1 mL/L的微量元素,自然pH值,所述微量元素由如下組分組成:5 g/L的FeSO4·7H2O;1.6 g/L的MnSO4·4H2O,1.4 g/L的ZnSO4·7H2O和20 g/L的CoCl2·6H2O。
1.2 實驗方法
1.2.1 產纖維素酶菌株的分離在超凈工作臺中,用滅菌的接種針挑取活體蝗蟲的腸道,放入到添加有適量生理鹽水和10個玻璃珠的無菌三角錐形瓶中,充分振蕩混勻。取上述菌液以10倍梯度進行稀釋。每個梯度各取100 μL菌液加入到CMC-Na初篩平板中,用玻璃涂抹棒將其涂布均勻,待平板上長出大小合適的菌落。
1.2.2 產纖維素酶菌株的篩選將不同菌落分別在CMC-Na復篩培養基上劃線,待平板上長出大小合適的單個菌落時,將不同的菌落按10倍梯度進行稀釋,每個梯度各取100 μL菌液,分別加入到剛果紅鑒別培養基中,28℃培養3 d,把菌體進行刮離,加入質量濃度為1 g/L的剛果紅染液染色10~15 min,無菌生理鹽水沖洗2~3次,選出能明顯產生透明圈的菌株。在平板上用游標卡尺分別測量菌株產生的透明圈直徑(D)和菌株的菌落直徑(d),并計算出每株菌株的菌徑比值H,挑取菌徑比值較大的5株菌落反復在固體培養基上進行劃線,直至得到純種單菌落。
透明圈的大小不能確切反映菌株的實際產酶能力,β-葡萄糖苷酶在纖維素降解過程中起至關重要的作用,在篩選出透明圈直徑與菌落直徑的比值H(D/d)較大的基礎上,進一步篩選出β-葡萄糖苷酶酶活較高的菌株[10],其總酶活(濾紙酶活)相對較高,于是將篩選出的上述5株菌株在七葉苷培養基上培養[11],進一步篩選,選擇在七葉苷平板上生長顏色相對較深的菌株進行保存。
1.2.3 孢子懸液的制備取一個在PDA平板上生長5 d的菌株F21平板,在超凈工作臺中向培養皿中倒入適量的無菌水,用三角玻璃涂布棒反復刮式,將孢子刮下,震蕩,三角漏斗8層紗布進行過濾,血球計數板顯微鏡下統計孢子個數,無菌水稀釋,調整到所需濃度106/mL,4℃冰箱保存備用[12]。
1.2.4 菌株生長曲線的繪制取1 mL菌株F21的孢子懸液,接種于含有50 mL種子液的250 mL三角錐形瓶中,置于溫度為28℃,轉速為200 r/min的搖床中振蕩培養,共接種33瓶,每天取出3瓶,在真空干燥箱烘干至恒重,稱量后取平均值,連續稱取11 d,以培養時間天數(d)為橫坐標,菌體的干重(mg)為縱坐標,繪制菌株F21的生長曲線圖。
1.2.5 粗酶液的制備取1 mL菌株F21新鮮孢子懸液,接種于含有50 mL種子液的250 mL三角錐形瓶中,置于溫度為28℃,轉速為200 r/min搖床中培養3 d,作為發酵種子液,將發酵種子液按5%體積比的接種量,接種于含有50 mL發酵液的250 mL三角瓶錐形中,28℃、200 r/min搖瓶發酵12 d,每天吸取1.0 mL發酵液,13 000 r/min離心時間2 min,去除沉淀,留取上清液,上清液即為粗酶液。取上清液用于酶活測定。
1.2.6 酶活力測定濾紙酶活力測定參照國家輕工行業標準QB 2583—2003的方法,以Whatman 1號濾紙為反應底物[13]。羧甲基纖維素酶活力測定參照國家輕工行業標準QB 2583—2003的方法[14],以質量濃度2 g/L CMC-Na溶液為反應底物。β-葡萄糖苷酶酶活力測定參照參考文獻的方法[11],略有改動,以1 mmol/L pNPG(4-硝基苯基-D-吡喃葡糖苷)溶液為反應底物。濾紙酶活力和羧甲基纖維素酶活力的酶活計算:以1 min催化底物生成1 μmol葡萄糖所需的酶量定義為一個酶活力單位(IU/mL)。β-葡萄糖苷酶活力的酶活計算:單位體積酶反應液每分鐘轉化相應底物釋放1 μmol產物所需的酶量為一個酶活力單位(IU/mL)。
葡萄糖標準曲線的繪制:采用紫外光譜法,配制10 mg/mL葡萄糖標準液,按一定比例稀釋后,反應體系中包含1.5 mL檸檬酸緩沖(0.5 mol/L,pH值4.8)、0.5 mL按一定比例稀釋后的葡萄糖標準液,加入3 mL DNS試劑沸水浴10 min后取出,快速冷卻至室溫后,蒸餾水定容至25 mL,吹打混勻。用10 mm比色皿,分光光度計在540 nm波長處測量吸光值。以葡萄糖量為橫坐標,以吸光值為縱坐標,繪葡萄糖制標準曲線,獲得線性回歸方程,y=0.228 23x-0.137 18,R2=0.997
β-葡萄糖苷酶酶活標準曲線的繪制:稱取139.1 mg 4-硝基苯酚,先加入適量的蒸餾水至完全溶解,然后定容至100 mL,配制成10 mmol/L的貯備液;用檸檬酸緩沖液(0.5 mol/L,pH值4.8)稀釋上述貯備液至1 mmol/L的標準貯備液;按不同濃度,將標準貯備液配制成濃度不同的標準使用液;按β-葡萄糖苷酶酶活測定的方法,測定不同濃度標準使用液對應的吸光值;以標準使用液的濃度為橫坐標,對應的吸光值為縱坐標,繪制標準曲線y=1.169 67x-0.007 95,R2=0.999
1.2.7 菌株分子生物學鑒定刮取新活化菌株F21的菌絲,利用酚氯仿法提取菌株的基因組,以菌株的DNA作為模板,采用真菌的通用引物進行PCR擴 增[15-16]。上 游 通 用 引 物ITS1(5′-tccgtaggtgaacctgcgg-3′),下 游 通 用 引 物ITS4(5′-tcctccgcttattgatatgc-3′)購于武漢天一華煜基因科技術有限公司。PCR反應體系(25 μL):2×Es Taq Master Mix(北京天根科技)12.5 μL、菌株F21的DNA模 板1 μL、上游引 物ITS1 1 μL、下游引物ITS4 1 μL、dd H2O 9.5 μL,對照組以加入ddH2O代替DNA模板。PCR擴增條件設定為:94℃,預變性5 min;94℃,變性30 s;56℃,退火30 s;72℃,延伸1 min,35個循環;72℃,延伸10 min,8℃,無窮。PCR擴增出的產物經質量分數為1.2%的瓊脂糖凝膠電泳進行檢測,其中,Marker DL 15000購自Takara公司。將PCR擴增產物膠回收后送至武漢天一華煜基因科技術有限公司進行測序,得到一條長631 bp的序列,在NCBI上進行BLAST相似性搜索,獲取近似典型菌株基因序列,進行序列比對,同時使用MEGA 7.0軟件繪制系統發育進化樹[17-19]。
2 結果與討論
2.1 高產纖維素酶活性菌株
采用剛果紅染色法從采樣的蝗蟲體內中共分離獲得5株在CMC-Na平板培養基上生長良好的菌株,透明圈直徑與菌落圈直徑之比H(D/d)及在七葉苷培養基上各菌株平板顏色深度見表1。剛果紅平板上菌徑比值越大,在一定程度上,反映出菌株內切葡聚糖酶活越高;七葉苷平板上產酶圈顏色越深,在一定程度上,反映出菌株β-葡萄糖苷酶酶活越高。菌株F21的菌徑比H值為2.83,相較于其他4株菌株比值略大,同時在七葉苷培養基上其培養基顏色呈紅棕色,相較于其他四株菌株顏色較深,最終選擇菌徑比值較大,培養基顏色較深的菌株F21進行保存,同時進行了菌種保藏。之后把菌株F21選擇為研究對象,進行后續實驗。
2.2 菌株的形態學特征及生長特性
(1)菌株F21在固體培養基平板上的菌落形態(圖1):菌株F21在培養到第4 d時菌落菌絲擴展較快,第5 d菌株就鋪滿培養皿。菌落特征為圓形,菌落邊緣輪紋狀,中央為乳白色的棉絮狀。
圖1 菌株F21的菌落形態Fig.1 Colony morphology of strain F21
(2)菌株F21長有菌絲,菌絲發達,呈放射狀生長,有孢子,在40×顯微鏡下觀察孢子短棒形(圖2)。

圖2 顯微鏡下菌株F21的孢子形態Fig.2 Spore morphology of strain F21 under microscope
(3)菌株F21在液體發酵培養基中培養數天發酵液變得澄清,搖床培養過程中,菌絲抱團形成菌絲球。
(4)菌株F21生長適溫25~28℃,pH值為6.0~7.0,好氧。
2.3 菌株的生長曲線
通過菌株F21的生長曲線(圖3)發現菌株F21在種子培養基中培養到第3 d到第4 d時,其干重增加明顯,說明其進入快速生長期,這與在平板上觀察到的現象較一致,但比平板上較早1 d進入快速生長期,可能與種子培養基的成分更適合菌株生長有關。所以在后續的實驗中取培養3 d的種子液按比例接種于發酵培養基中。
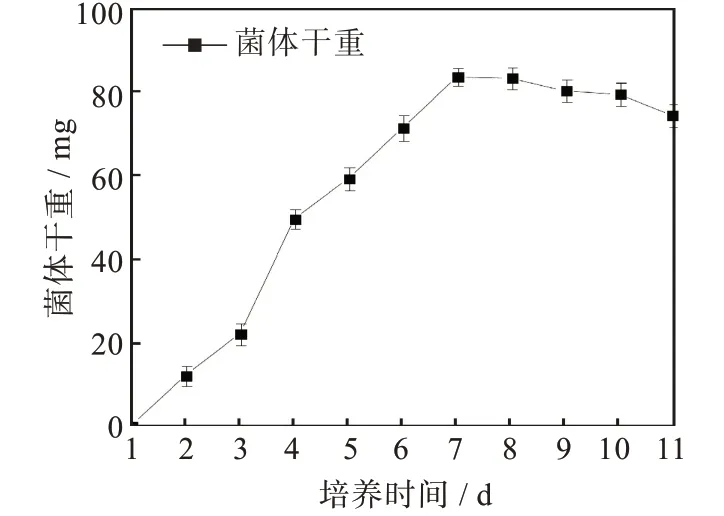
圖3 菌株F21的生長曲線Fig.3 Growth curve of strain F21
2.4 菌株產纖維素酶活力
濾紙酶活性可代表纖維素酶對纖維素的整體水解能力,進而結合羧甲基纖維素酶活性和β-葡萄糖苷酶活性大小對菌株F21進行酶活力評價。共搖瓶發酵培養12 d,間隔24 h取樣,制備粗酶液,測定菌株F21的酶活力。
菌株F21產生的濾紙酶活性(圖4)在發酵培養第3 d出現活力高峰達0.212 IU/mL,羧甲基纖維素酶活性在發酵培養第4 d時達峰值1.593 IU/mL,β-葡萄糖苷酶酶活性(圖5)從第2 d開始升高,在第6 d時達到峰值39.069 IU/mL,隨之又逐漸下降。
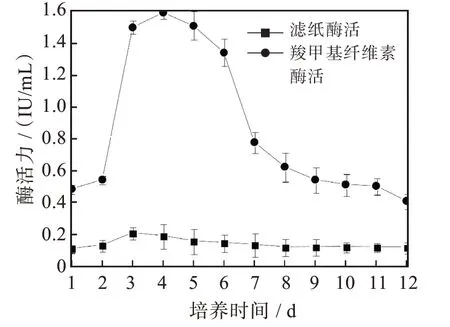
圖4 菌株F21的濾紙酶活性和羧甲基纖維素酶活性曲線Fig.4 Filter paper enzyme activity and carboxymethyl cellulase activity of strain F21
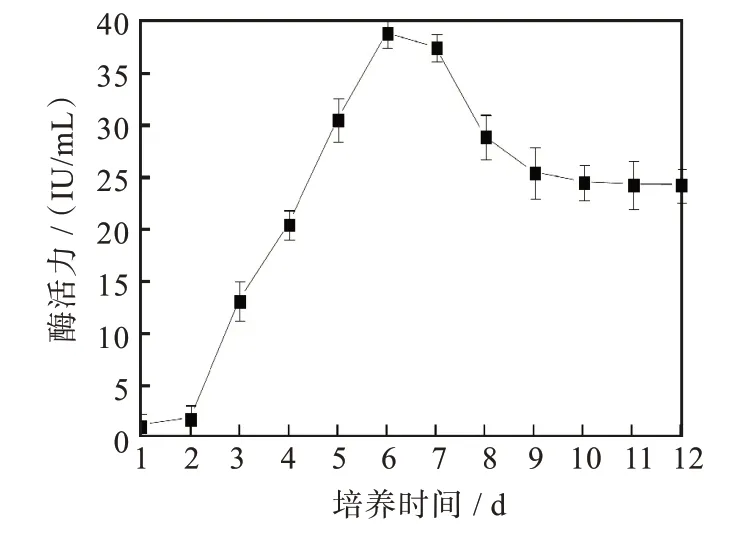
圖5 菌株F21的β-葡萄糖苷酶活性曲線Fig.5 β-glucosidase activity of strain F21
2.5 菌株F21的18S rDNA序列分析
以菌株F21的基因組DNA為模板,采用真菌通 用 引 物ITS1/ITS4進 行PCR擴 增,PCR產 物(圖6)經測序得到一條長度為631 bp的片段。將片段測序結果在NCBI數據庫進行BLAST比對,結果顯示與GenBank登錄號為MT644869.1的煙管菌(Bjerkandera adusta)相似比例為99.83%。因此,菌株F21在系統發育分類學上屬于煙管菌。綜合菌株F21的菌落形態和分子生物學鑒定測序結果,確定菌株F21為煙管菌屬真菌菌株,命名為Bjerkandera adustaF21。同時將該序列與BlastN比對結果中來源不同的Bjerkandera adusta序列以及同屬不同種的ITS序列進行多序列比對分析,利用MEGA7.0軟件制作菌株F21系統發育樹(圖7)。
圖6 菌株F21的PCR產物圖Fig.6 PCR product graph of strain F21

圖7 菌株F21的系統發育樹Fig.7 Phylogenetic tree of strain F21
3 結論
本試驗從野外采集到的蝗蟲腸道內分離篩選出一株產纖維素酶酶活較高的真菌菌株F21,通過形態學和分子生物學鑒定,確定菌株F21屬于真菌中的煙管菌(Bjerkandera adusta)。對菌株F21在液體發酵培養基中不同階段纖維素酶活力測定,結果表明其濾紙酶活最高為0.212 IU/mL,羧甲基纖維素酶活最高為1.593 IU/mL,β-葡萄糖苷酶活最高為39.069 IU/mL。為實現對纖維素資源的高效利用[20],該菌株后續還需要馴化、誘變和基因工程改造等手段提高其纖維素酶活性。
