貝母屬植物葉綠體基因組結構與系統發育的關系
2023-01-12 06:03:12馬曉君郭宗雷王信宏玄靜田靜劉歡隋園園束靖
山東農業大學學報(自然科學版) 2022年5期
馬曉君,郭宗雷,王信宏,玄靜,田靜,劉歡,隋園園,束靖
貝母屬植物葉綠體基因組結構與系統發育的關系
馬曉君,郭宗雷,王信宏,玄靜,田靜,劉歡,隋園園,束靖*
山東農業工程學院, 山東 濟南 250000
貝母屬植物形態相似,難以區分。通過對葉綠體基因組進行分析,可以為貝母屬植物的系統發育、分子鑒定和開發利用提供理論依據。我們基于已發表的貝母屬物種的葉綠體基因組序列,利用相關生物信息學方法對其葉綠體基因組進行分析。結果顯示,貝母屬29個物種葉綠體基因組大小在151058~152434 bp。基因數量為128~133個,rRNA編碼基因數目穩定,蛋白質編碼基因和tRNA編碼基因存在差異。四分區分析發現,IRb/SSC邊界存在明顯差異。串聯重復和單核苷酸重復含量最豐富。共線性分析未檢測到基因重排,其葉綠體基因組高度保守。基于葉綠體基因組的系統發育分析表明,貝母屬植物分為4個主要分支。對貝母葉綠體基因組進行核苷酸多態性分析,發現IR比單拷貝區更保守,非編碼區的突變頻率比編碼區高。
貝母屬植物; 葉綠體; 基因; 系統發育
中藥材“貝母”屬百合科(Liliaceae)貝母屬(),絕大多數是多年生草本植物。入藥部分為的鱗莖,具有清熱潤肺、化痰止咳等功效[1]。近年來,多位學者已經對國產貝母屬的植物進行了系統分類研究,發表了許多新種和新變種,使國產貝母屬植物“種”數已達80個,“變種”名稱數已達52個[2]。由于貝母屬植物在形態上十分相近,難以在形態上進行區分,在中藥材市場中常出現以次充好、以偽充真等問題[3]。
被子植物葉綠體通常具有由一對倒置重復(inverted repeat, IR)區,小單拷貝(small single-copy, SSC)區,大單拷貝(large single-copy, LSC)區組成的四分體結構。眾多研究表明,葉綠體基因組結構和序列保守,在分子標記開發、遺傳育種、物種鑒定及系統進化研究等方面具有重要作用[4, 5]。尤其是隨著DNA測序技術的快速發展,越來越多地物種完成葉綠體基因組測序[6]。并且葉綠體基因組相較于DNA條形碼具有更高的分辨率,已成為中草藥鑒定、分類的重要工具[7]。本試驗基于已發表的貝母屬葉綠體基因組序列,利用生物信息學方法對其葉綠體基因組的特征、基因數量與長度、結構變異、重復序列、基因重排、系統發育和序列多態性進行分析,為貝母屬植物的系統發育、分子鑒定和開發利用提供理論依據。
1 數據收集和研究方法
1.1 數據收集
在Genbank數據庫(https://www.ncbi.nlm.nih.gov/genome/organelle/)下載已發表的29個百合科貝母屬植物的葉綠體基因組序列信息,收集其物種名稱、GenBank登錄號等(見表1)。
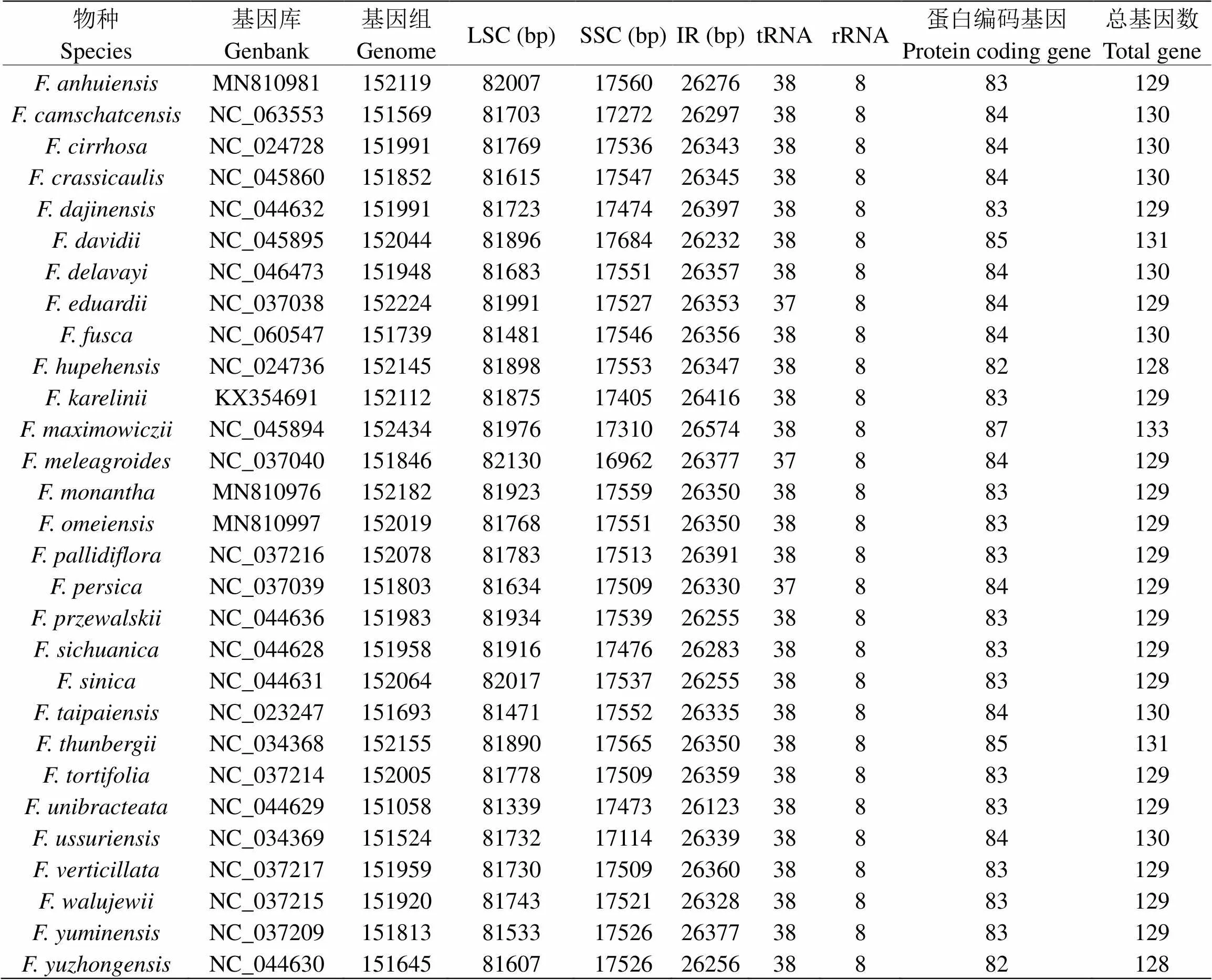
表1 29種貝母屬植物葉綠體基因組注釋信息
1.2 葉綠體基因組注釋
利用在線網站CPGAVAS2(http://47.96.249.172:16019/analyzer/home)進行葉綠體基因組的注釋[8]。Excel統計葉綠體基因組長度、基因數量、四分區長度等信息。
1.3 葉綠體基因組比較
通過IRSCOPE(https://irscope.shinyapps.io/irapp/)對葉綠體基因組四分區進行可視化[9]。以葉綠體基因組作為參考序列,通過在線網站mVISTA(http://genome.lbl.gov/vista/mvista/submit.shtml)中的Shuffle-Lagan模式進行全基因組比較[10];利用Mauve軟件對葉綠體基因組進行全局比對,并進行共線性分析[11]。
1.4 SSR與重復序列分析
通過在線分析網站REPuter(https://bibiserv.cebitec.uni-bielefeld.de/reputer/)查找散在重復序列和回文重復序列[12],參數設置為:漢明距離3,最大計算重復次數90,最小重復單元長度30。串聯序列通過TRF(Tandem Repeats Finder)(http://tandem.bu.edu/trf/trf.html)查找[13],使用默認參數。簡單重復序列(Simple Sequence Repeats, SSRs)通過MISA網站(https://webblast.ipk-gatersleben.de/misa/)查找[14],重復次數閾值設置為:單核苷酸10、二核苷酸5、三核苷酸4、四核苷酸3、五核苷酸3、六核苷酸3。
2.5 構建系統發育樹
本研究中29個物種的葉綠體基因組長度和四分區長度差異不大,選擇葉綠體全基因組數據進行分析,通過最大似然法(maximum likelihood, ML)構建系統發育樹。使用百合科百合屬的、、作為外類群[15],與貝母屬一起構建系統發育樹。使用MAFFT軟件對葉綠體基因組進行多序列比對[16],通過MEGA獲得最佳替代模型并構建ML系統發育樹[17],bootstrap次數為1000次。
2.6 序列多態性分析
使用MAFFT進行多序列比對[16],將比對結果導入DNAsp[18],計算葉綠體基因組序列的核苷酸多態性(pi),參數設置為:window length 600,step size 200。
2 結果與分析
2.1 葉綠體基因組結構特征
通過葉綠體基因組注釋信息(見表1)發現,本研究中29個物種的葉綠體基因組總長度差距不大,最長的長度為152434 bp,最短的長度為151058 bp,相差約1.4 kb。葉綠體基因組包括一對反向重復區(IR)、大單拷貝區(LSC)和小單拷貝區(SSC)。本研究中LSC區長度在81339~82130 bp之間,最長的為,最短的是,相差791 bp;SSC區長度在16962~17684 bp之間,最長的為,最短的為,相差722 bp;兩個IR區域等長,在26123~26574 bp之間,其中長度最長,長度最短,相差451 bp。
2.2 基因數量
29個百合科貝母屬植物的葉綠體基因總數在128~133之間(表1),其中17個物種的基因數量為129,占大多數。rRNA編碼基因數目穩定,全部為8個;tRAN數大多數為38,只有3個物種含有37個tRNA;蛋白質編碼基因數量在82~87之間,其中14個物種含有83個(見表1),可見基因總數差異主要是由蛋白編碼基因差異引起的。
2.3 四分區邊界分析
葉綠體四分區邊界為LSC/IRb、IRb/SSC、SSC/IRa、IRa/LSC。通過分析各邊界葉綠體基因的分布狀況,可以分析貝母葉綠體在進化過程中IR區收縮與擴張現象。如圖1可以得知,LSC/IRb邊界位于和基因內,由于IR區域的收縮和擴張,有5個物種的全部位于LSC區域;中的在邊界幾乎均勻分布;其余23個物種的有236~268 bp位于LSC區域,11~43 bp進入IRb區域。IRb/SSC邊界處、基因分布存在較大差異,有16個物種的全部位于IRb區內,其中的長度更短;12個物種的大部分位于IRb區,16~121 bp進入SSC區域;的基因丟失;大多數物種位于SSC區內,5個物種的跨過IRb/SSC邊界,有8~33 bp位于IRb區。SSC/IRa邊界處長度與分布大致相似,均為4245~4394 bp位于SSC區,1147~1263 bp位于IRa區;但和的長度較短且位于SSC區的長度發生變化。IRa/LSC邊界位于和之間,只有的IRa/LSC邊界位于基因內。

圖1 29個貝母屬植物葉綠體基因組四分區邊界比較
2.4 重復序列及SSR分析
在貝母屬29個物種中共發現1853個重復序列,包括28.87%散在重復、33.51%回文重復和37.61%串聯重復(圖2A,B)。單個物種的重復序列數目大多在57~67之間,但僅含有51個,4個物種的重復序列數超過70個。SSR共發現2176個。其中單核苷酸數目最多,占67.69%(1473個),二核苷酸占17.88%(389個),三核苷酸占3.08%(67個),四核苷酸占10.11%(220個),五核苷酸占1.19%(26個),僅有含有1個六核苷酸(圖2C,D)。29個物種中,SSR總數在59~83之間,其中數目最少,數目最多。單核苷酸數為39~58;二核苷酸數為9~16;三核苷酸數為1~6;四核苷酸數為6~9;五核苷酸數目為0~3之間,其中9種貝母不含五核苷酸重復。
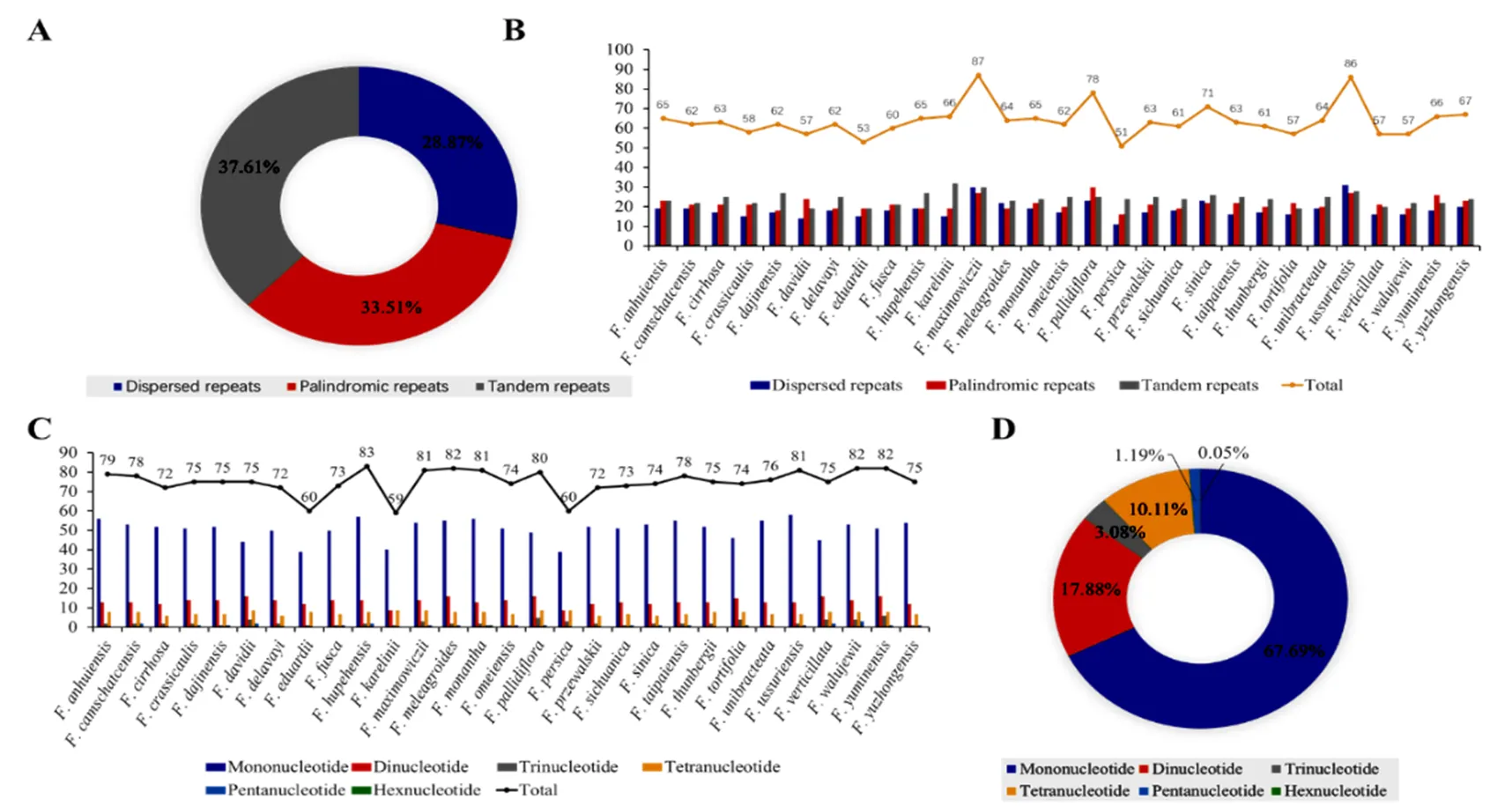
(A)3種重復類型的比例The ratio of three repeated types;(B)3種重復類型的數目The number of three repeats types;(C)不同類型SSR的比例The ratio of different SSR;(D)不同類型SSR的數目The number of different SSR
2.5 葉綠體基因組水平對比和共線性分析
全基因組比對結果表明(圖3),整個葉綠體基因組高度保守,且IR比單拷貝區更保守,非編碼區的突變頻率比編碼區高。、、基因的編碼區比其他基因的編碼區更具可變性。共線性分析結果顯示沒有檢測到大片斷基因重排現象(圖4),也表明貝母屬的29個物種相對保守。
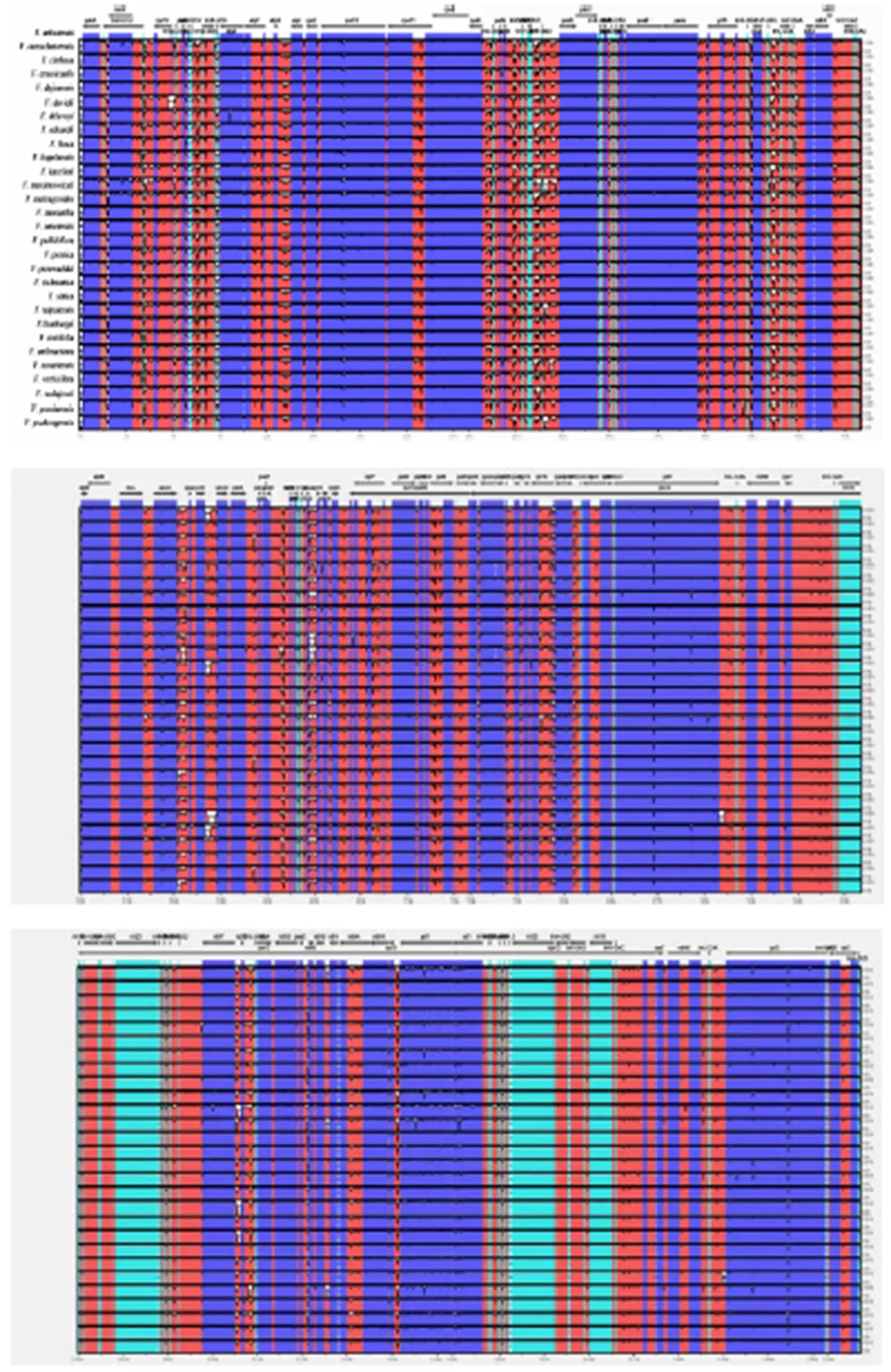
圖3 29種貝母屬植物葉綠體基因組比對
注:軸代表葉綠體基因組中的坐標,軸表示對齊區域的序列相似性在50%~100%。箭頭表示基因及轉錄方向。
Note: The x-axis represents the coordinate in the chloroplast genome. The y-axis indicates that the sequence similarity of the aligned region is between 50% and 100%. Arrows indicate genes and transcriptional direction.

圖4 29種貝母屬植物葉綠體基因組共線性分析
2.6 系統發育樹分析
使用ML法對29種貝母屬植物及3種百合科植物的葉綠體全基因組序列進行系統發育樹分析,最佳替代模型為GTR+G+I。進化樹各節點分支分辨率高(圖5),位于進化樹基部,屬于較早分化的類群,且與百合屬聚類為一支;是單獨的一支,與其他物種為姐妹類群;、、、聚為一支;與聚為一支;、、、、聚為一支;其余16種貝母聚類為一支。
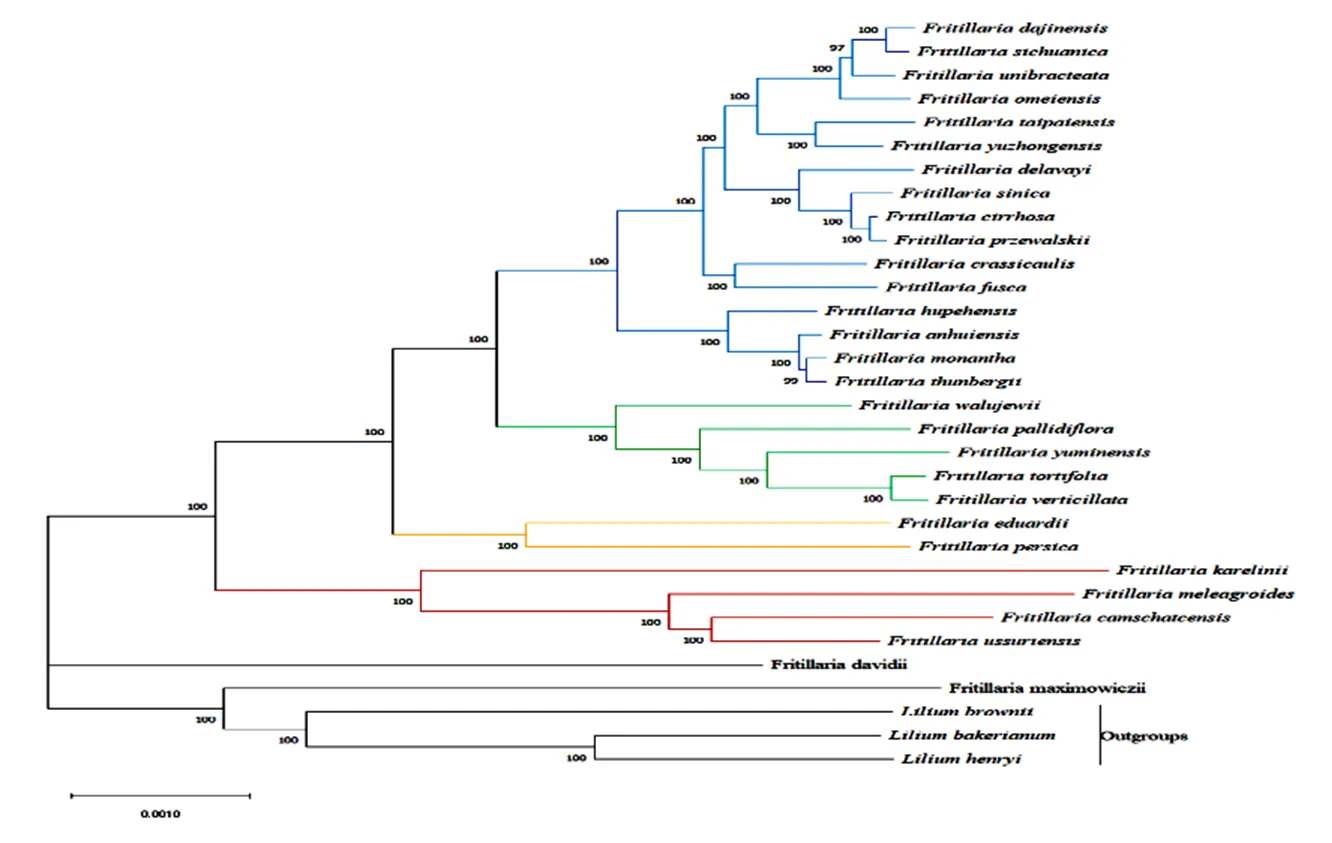
圖5 基于葉綠體全基因組序列構建的ML樹
2.7 序列多態性分析
共檢測到718個核苷酸多態性位點,核苷酸多態性范圍為0~0.02176(圖6),平均值為0.005254248,-、-、-等基因或基因間隔區具有較高的變異,它們的核苷酸多態性均大于0.02,其中-的多態性最高,為0.02176,-和-的多態性分別為0.02106和0.02033。
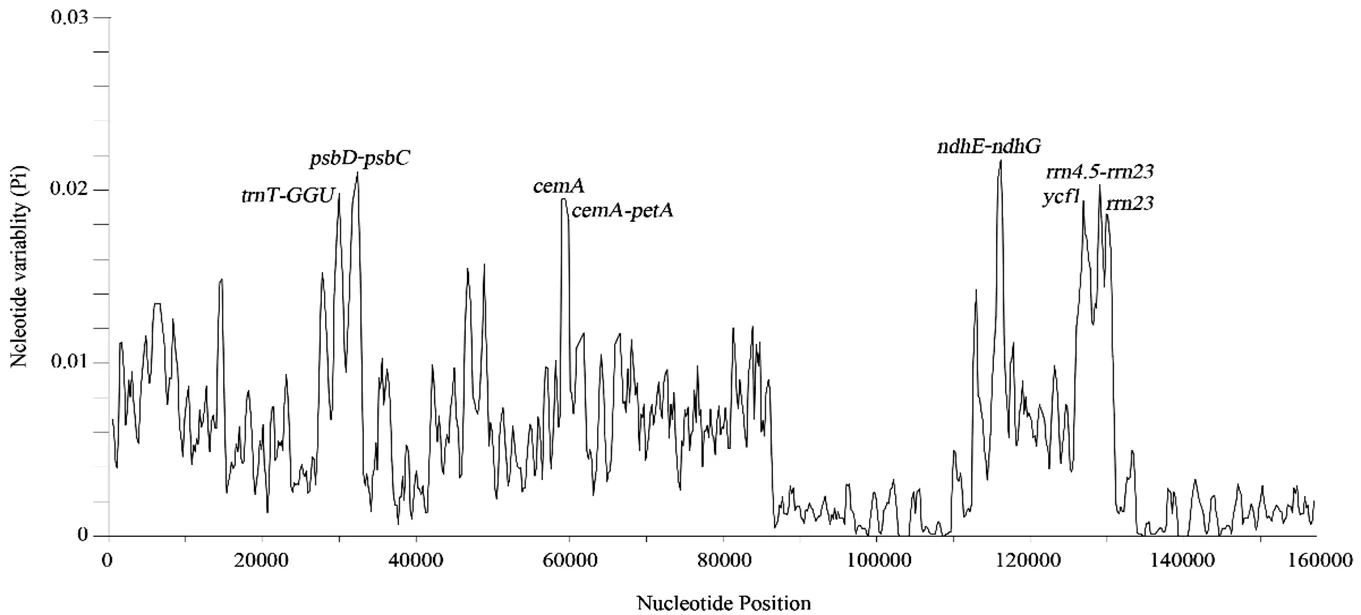
圖6 29種貝母屬植物葉綠體基因組滑動窗口分析
3 討論與結論
貝母屬29個物種葉綠體基因組的大小在152434~151058 bp之間,LSC區、SSC區、IR區長度與被子植物葉綠體基因組長度相吻合[19]。本研究中,各物種間基因組大小相差約1.4 kb左右,LSC、SSC以及IR區域相差分別為0.8 kb、0.7 kb以及0.45 kb左右。葉綠體基因組IR區的收縮與擴張普遍發生在被子植物進化過程中[20],是基因組結構變異的重要原因。貝母屬葉綠體基因組結構、基因數量和順序高度保守[21]。29種貝母屬植物的4個邊界區分別跨過、-、、-。但IRb/SSC邊界處、的分布在不同物種中存在一定差異,貝母屬葉綠體基因組長度變異可能是IR和SSC邊界變化引起的[22]。與百合科植物類似,貝母屬植物葉綠體基因組中的LSC/IRb邊界大多擴展到基因,這可能是百合科貝母屬植物的共同祖征[23,24]。
葉綠體共線性分析沒有檢測到基因重排現象,全基因組序列比對也顯示葉綠體基因組序列相似度很高,其IR區比單拷貝區更保守,非編碼區的突變頻率比編碼區高,與前人研究結果一致[21,25]。長重復序列與SSR在葉綠體基因組結構變異中具有重要作用,常用于藥用植物物種鑒定、群體遺傳學研究[26,27]。在29種貝母屬植物葉綠體基因組中單核苷酸數目最多,因此單核苷酸可能比其他類型的SSR對貝母遺傳變異的貢獻更大。有研究表明、-、能更好地識別分辨貝母屬植物[28],但在本研究中-、的pi值不高,的pi值為0.01938。也被認為是陸地植物最有可能的葉綠體DNA條形碼[29],有望于開發成為高效鑒別貝母屬植物DNA條形碼。
本研究構建的構建ML樹中,大部分系統發育關系與Zhang T、Chen Q等的結果相似[27,28],除與外,主要聚為4個主要分支,與百合屬聚類為一支,并且分辨率達到100 BP而非Zhang T等的75 BP[27]。其余不同可能是因為所使用的葉綠體基因組數據不同導致。
[1] 徐順連,曾中蘭,林青,等.貝母化學成分及藥理作用的研究[J].青海草業,2021,30(1):43-46
[2] 肖培根,姜艷,李萍,等.中藥貝母的基原植物和藥用親緣學的研究[J].植物分類學報,2007(4):473-487
[3] 羅焜,馬培,姚輝,等.基于ITS2序列鑒定川貝母及其混偽品基原植物[J].世界科學技術(中醫藥現代化),2012,14(1):1153-1158
[4] Li HT, Yi TS, Gao LM,. Origin of angiosperms and the puzzle of the Jurassic gap [J]. Nat Plants, 2019,5(5):461-70
[5] Nie Y, Foster CSP, Zhu T,Accounting for uncertainty in the evolutionary timescale of green plants through clock-partitioning and fossil calibration strategies [J]. Syst Biol, 2020,69(1):1-16
[6] Dobrogojski J, Adamiec M, Luciński R. The chloroplast genome: a review [J]. Acta Physiologiae Plantarum, 2020,42(6):98
[7] 林小涵,劉志華,李卿,等.藥用植物葉綠體基因組研究[J].世界科學技術(中醫藥現代化),2010,12(3):442-446
[8] Shi L, Chen H, Jiang M,. CPGAVAS2, an integrated plastome sequence annotator and analyzer [J]. Nucleic Acids Res, 2019,47(1):65-73
[9] Amiryousefi A, Hyvonen J, Poczai P. IRscope: an online program to visualize the junction sites of chloroplast genomes [J]. Bioinformatics, 2018,34(17):3030-3031
[10] Frazer KA, Pachter L, Poliakov A,. VISTA: computational tools for comparative genomics [J]. Nucleic Acids Res, 2004,32:273-279
[11] Darling AC, Mau B, Blattner FR,. Mauve: multiple alignment of conserved genomic sequence with rearrangements [J]. Genome Res, 2004,14(7):1394-1403
[12] Kurtz S, Choudhuri JV, Ohlebusch E,. REPuter: the manifold applications of repeat analysis on a genomic scale [J]. Nucleic Acids Res, 2001,29(22):4633-4642
[13] Benson G. Tandem repeats finder: a program to analyze DNA sequences [J]. Nucleic Acids Res, 1999,27(2):573-580
[14] Beier S, Thiel T, Munch T,. MISA-web: a web server for microsatellite prediction [J]. Bioinformatics, 2017,33(16):2583-2585
[15] Li Y, Zhang Z, Lv G. The complete chloroplast genome of, a rare and endangered species endemic to China [J]. Mitochondrial DNA B Resour, 2017,2(2):913-914
[16] Katoh K, Misawa K, Kuma K,. MAFFT: a novel method for rapid multiple sequence alignment based on fast fourier transform [J]. Nucleic Acids Res, 2002,30(14):3059-3066
[17] Tamura K, Stecher G, Kumar S. MEGA11: Molecular evolutionary genetics analysis version 11 [J]. Mol Biol Evol, 2021,38(7):3022-3027
[18] Rozas J, Ferrer-Mata A, Sanchez-Delbarrio JC,. DnaSP 6: DNA sequence polymorphism analysis of large data sets [J]. Mol Biol Evol, 2017,34(12):3299-3302
[19] Zhang T, Fang Y, Wang X,. The complete chloroplast and mitochondrial genome sequences of: insights into the evolution of plant organellar genomes [J]. PLoS One, 2012,7(1):e30531
[20] Fu CN, Li HT, Milne R,. Comparative analyses of plastid genomes from fourteen Cornales species: inferences for phylogenetic relationships and genome evolution [J]. BMC Genomics, 2017,18(1):956
[21] Huang J, Yu Y, Liu YM,. Comparative chloroplast genomics of(liliaceae), inferences for phylogenetic relationships betweenandand plastome evolution [J]. Plants, 2020,9(2):133-148
[22] Bi Y, Zhang MF, Xue J,. Chloroplast genomic resources for phylogeny and DNA barcoding: a case study on Fritillaria [J]. Sci Rep, 2018,8(1):1184
[23] Li P, Lu RS, Xu WQ,. Comparative genomics and phylogenomics of east asian tulips (Amana, Liliaceae) [J]. Front Plant Sci, 2017,8:451
[24] Li Q, Li Y, Song J,. High-accuracy de novo assembly and SNP detection of chloroplast genomes using a SMRT circular consensus sequencing strategy [J]. New Phytologist, 2014,204(4):1041-1049
[25] Park I, Kim WJ, Yeo SM,. The complete chloroplast genome sequences ofmaxim. andD. don, and comparative analysis with otherspecies [J]. Molecules, 2017,22(6):982
[26] 田星,劉瑩瑩,張穎敏,等.藜蘆屬藥用植物的葉綠體基因組比較分析和系統發育研究[J].中草藥,2022,53(4):1127-1137
[27] Zhang T, Huang S, Song S,. Identification of evolutionary relationships and DNA markers in the medicinally important genusbased on chloroplast genomics [J]. PeerJ, 2021,9:e12612
[28] Chen Q, Hu H, Zhang D. DNA Barcoding and phylogenomic analysis of the genusin China based on complete chloroplast genomes [J]. Front Plant Sci, 2022,13:764255
[29] Dong W, Xu C, Li C,. ycf1, the most promising plastid DNA barcode of land plants [J]. Sci Rep, 2015,5:8348
Relationship between Chloroplast Genomes Structures fromPlantsand Phylogeny
MA Xiao-jun, GUO Zong-lei, WANG Xin-hong, XUAN Jing, TIAN Jing, LIU Huan, SUI Yuan-yuan, SHU Jing*
,250000,
It is difficult to distinguish between plants of thebecause of their morphological similarity. Characteristics and phylogeny of chloroplast genomes, can provide a theoretical basis for study of the phylogeny, molecular identification, and resource exploitation of the. Based on the published chloroplast genome sequences of 29species, their chloroplast genomes were analyzed using bioinformatics methods. The results showed that, the chloroplast genome is between 151058 bp and 152434 bp. There are 128 to 133 genes in the, with a stable number of rRNA-coding genes, and differences in protein-encoding and tRNA-encoding genes. Significant differences exist at the IRb/SSC boundary. The highest content of tandem repeats and mononucleotide. No gene rearrangements were detected by covariance analysis, reflecting the high degree of conservation in the chloroplast genome of the. Phylogenetic analysis based on the chloroplast genome indicates that thecan be divided into four major phylogenetic clades. Nucleotide polymorphism analysis of the chloroplast genome offound that the IR was more conserved than the single-copy regions, and the non-coding region was mutated more frequently than the coding region.
; chloroplast; gene; phylogeny
Q75
A
1000-2324(2022)05-0811-08
2022-09-24
2022-10-11
國家重點研發計劃(2019YFD1000102);山東省良種工程(2019LZGC009);山東農業工程學院校企科研創新團隊(YBZR202204)
馬曉君(1997-),女,研究生,助教,研究方向:基因工程. E-mail:mxjun7@163.com
通訊作者:Author for correspondence. E-mail:shujing79@163.com
猜你喜歡
課堂內外·初中版(科學少年)(2025年1期)2025-02-28 00:00:00
課堂內外·初中版(科學少年)(2025年2期)2025-02-28 00:00:00
英語世界(2023年10期)2023-11-17 09:18:18
科學大眾(中學)(2019年3期)2019-05-17 10:04:30
汽車觀察(2018年10期)2018-11-06 07:05:26
少兒科學周刊·兒童版(2017年5期)2017-06-29 22:24:28
少兒科學周刊·兒童版(2017年5期)2017-06-29 16:46:33
紅領巾·萌芽(2017年5期)2017-06-23 10:35:59
爆笑show(2016年7期)2017-02-09 09:36:13
少兒科學周刊·兒童版(2015年10期)2015-11-07 03:42:03
