水合氧化鋯吸附劑在電池級硫酸錳溶液中除氟研究
2023-01-18 05:36:06胡慧萍董和大彭奇凡
中南大學學報(自然科學版) 2022年11期
關鍵詞:質量
胡慧萍,董和大,彭奇凡
(1.中南大學 化學化工學院,湖南 長沙,410083;2.長沙華迪水處理技術有限公司,湖南 長沙,410083)
隨著煤、石油等天然資源的日益枯竭,以鋰離子電池為代表的新興能源逐漸占據能源市場主導地位[1-2]。電池級硫酸錳是用于制備鋰離子電池正極材料如鎳鈷錳三元材料、錳酸鋰材料的重要合成鹽。因此,硫酸錳的純度是決定鋰電池性能的關鍵因素之一。
目前,我國硫酸錳主要是通過將菱錳礦用硫酸浸取而得[3],獲得硫酸錳的同時引入了大量的鈣鎂離子。為除去鈣、鎂雜質,在工業生產中常采用加入過量MnF2的方式,使鈣、鎂離子形成難溶性CaF2和MgF2沉淀而除去。然而,過量MnF2的使用會引起MnSO4·H2O產品中氟含量過高的問題。研究表明,氟濃度過高不僅會腐蝕設備,還會降低錳酸鋰電池的電池容量與充放電效率[4],故需將氟含量控制在較低水平。目前,我國針對電池級硫酸錳產品中氟含量上限尚未做出具體要求,類比同樣可用作合成鋰離子電池正極材料的高純碳酸鋰,參照YS/T 546—2008 對高純碳酸鋰的要求標準,純度99.99%的Li2CO3中氟質量分數應小于5×10-5,電池級MnSO4·H2O 產品中氟質量分數也應以低于5×10-5為宜。以質量分數為20%的硫酸錳溶液結晶獲得電池級MnSO4·H2O固體產品為例,結晶過程相當于將氟濃縮了約5倍,為避免結晶過程中氟析出而使產品中氟含量大于5×10-5,應控制結晶前硫酸錳溶液中氟質量濃度低于10 mg/L。
已有的除氟研究主要是圍繞處理含氟廢水開展的,針對硫酸錳溶液中氟的去除鮮有報道。應用于含氟廢水的除氟方法主要有混凝法[5]、離子交換法[6]、化學沉淀法[7]、膜工藝法[8]等。混凝法雖然操作簡單,但需加入大量混凝劑,引起溶液pH下降,需重新加堿調高pH 才能使混凝劑沉淀完全,而且該法產泥量大,污泥中會夾帶大量錳,導致錳損失過大,不適用于深度除氟。離子交換法和膜工藝法除氟效果好,但成本過高。吸附法可較好地適用于電池級硫酸錳溶液中除氟,因其在控制除氟過程中不引入新雜質,可減少錳損失。用于電池級硫酸錳溶液除氟的吸附劑應具有吸附容量大、酸性條件下不溶解、合成簡單的特點。有學者采用硫酸鋁改性活性氧化鋁作為吸附劑脫除硫酸錳溶液中氟化物[9],吸附容量為1.96 mg/g,吸附容量偏低,在酸性硫酸錳溶液中,鋁可能會溶出而引進新雜質。
本文采用沉淀法制備水合氧化鋯吸附劑,以起始pH、吸附劑添加量、吸附時間、起始氟質量濃度、溫度為參數研究不同條件下吸附劑對硫酸錳溶液中氟的吸附效果的影響,考察除氟后錳損失與溶液中鋯殘留情況,并結合SEM,FT-IR 和XPS等表征技術就吸附機理進行分析。
1 實驗
1.1 主要試劑與儀器
氧氯化鋯(ZrOCl2·8H2O,分析純,上海麥克林生化試劑有限公司);氟化鈉(NaF,優級純,國藥集團化學試劑有限公司);硫酸錳(MnSO4·H2O,分析純,國藥集團化學試劑有限公司);其他試劑均為分析純,實驗用水為去離子水。實驗過程中使用的模擬含氟硫酸錳溶液由硫酸錳與氟化鈉混合溶解配制而成。
水浴恒溫振蕩器(THZ-82型,江蘇金壇市中大儀器廠);pH計(FE-20型,梅特勒-托利多儀器(上海)有限公司);離子計(PXSJ-216F型,儀電科學儀器股份有限公司,配PF-2-01 型氟離子選擇性電極);電熱恒溫鼓風干燥箱(DHG-9076A 型,上海精宏實驗設備有限公司);電感耦合等離子發射光譜儀(ICP,PerkinElmer Avio 500 型,德國珀金埃爾默股份有限公司)。
1.2 水合氧化鋯吸附劑合成
水合氧化鋯吸附劑在DOU 等[10]的基礎上加以改進:稱取32.225 g ZrOCl2·8H2O溶于0.2 mol/L鹽酸中,加入25 mL 乙二醇,超聲分散10 min 后定容至500 mL。向上述鋯鹽溶液以900 r/min 的速度逐滴滴加4 mol/L 氨水至pH=7,繼續攪拌2 h,60 ℃靜置陳化24 h 后抽濾,再用去離子水、乙醇交替洗滌濾餅,最后將濾餅置于60 ℃恒溫干燥箱干燥24 h,研細備用。
1.3 吸附性能測試
1.3.1 起始pH影響
稱取0.1 g 水合氧化鋯加入錳質量濃度為90.39 g/L、氟質量濃度為50 mg/L 的50 mL 硫酸錳溶液中,用稀硫酸和MnCO3將溶液pH 分別調至3.5,4.0,4.5,5.0和5.5,20 ℃恒溫振蕩吸附8 h,再將其過濾,測定濾液中氟質量濃度。
1.3.2 吸附劑用量影響
分 別 稱 取0.02,0.04,0.10,0.16,0.24 和0.32 g水合氧化鋯加入錳質量濃度為90.39 g/L、氟質量濃度為50 mg/L的50 mL硫酸錳溶液中,用稀硫酸和MnCO3將溶液pH 分別調至4.0,20 ℃恒溫振蕩吸附8 h,過濾后測定濾液中氟質量濃度。
1.3.3 吸附動力學實驗
取0.1 g水合氧化鋯加入錳質量濃度為90.39 g/L、氟質量濃度50 mg/L的50 mL硫酸錳溶液中,20 ℃恒溫振蕩2 min后過濾。其他條件不變,只改變振蕩時間,測定不同吸附時間下濾液中氟質量濃度。
1.3.4 吸附等溫實驗
設置模擬含氟硫酸錳溶液起始氟質量濃度分別為20,50,100,140,200,250 和350 mg/L,用稀硫酸和MnCO3將溶液pH 均調至4.0,分別在5,20 和35 ℃下恒溫振蕩吸附8 h,過濾后測定濾液殘留的氟質量濃度。
1.3.5 吸附劑表征
吸附劑吸附前后表面形貌采用掃描電子顯微鏡(SEM,JSM-7610Fplus 型,日本)檢測;用傅里葉變換紅外光譜(FT-IR,Nicolet 6700 型,美國)研究吸附劑官能團變化;元素結合能變化采用X 射線光電子能譜(XPS,Thermo Scientific K-Alpha型,美國)分析,使用Thermo Avantage軟件對數據進行分析擬合。
1.3.6 檢測方法
采用氟離子選擇性電極法測試溶液中氟質量濃度,吸附劑的平衡吸附容量按下式計算:
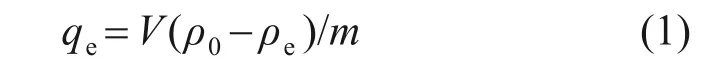
式中:ρ0為硫酸錳溶液中的起始氟質量濃度,mg/L;ρe為達到吸附平衡后溶液中的氟質量濃度,mg/L;qe為吸附劑平衡吸附容量,mg/g;V為待吸附溶液體積,L;m為吸附劑質量,g。
2 結果與討論
2.1 溶液起始pH的影響
溶液起始pH 對水合氧化鋯吸附劑在硫酸錳溶液中的氟離子吸附效果的影響見圖1。由圖1可知:當起始pH為3.5~5.5時,吸附劑對氟離子均有吸附效果。當起始pH 為4.0 時,吸附劑對氟離子有最佳吸附效果,此時溶液中氟質量濃度由50.00 mg/L降至8.54 mg/L,吸附劑的平衡吸附容量為20.73 mg/g。當起始pH 降至3.5 時,溶液殘留氟質量濃度升高至11.84 mg/L。當pH 大于4.0 時,pH越高,溶液中殘留氟質量濃度越高,吸附效果持續降低,當pH 為5.5 時,吸附劑的平衡吸附容量下降至15.04 mg/g。
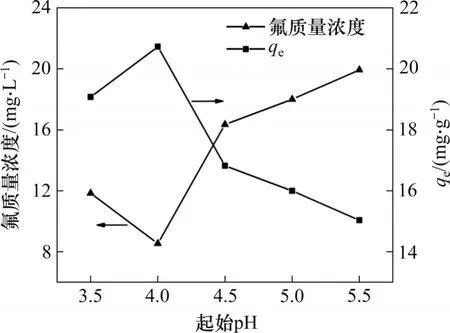
圖1 起始pH對吸附效果的影響Fig.1 Effect of initial pH on adsorption efficiency
當溶液pH 由4.0 降至3.5 時,氫離子濃度增大,氟離子與氫離子易結合形成HF,使吸附效果下降。當pH>4時,隨pH升高,OH-濃度增大,由于OH-與F-在離子半徑及電負性上具有相似性,OH-也會占據吸附活性位點[11],OH-與F-產生競爭吸附導致吸附效果下降。
2.2 吸附劑用量影響
為探究吸附劑的合理用量,在最少量的吸附劑使用條件下確保溶液中殘留氟質量濃度小于10 mg/L,研究吸附劑用量對吸附效果的影響,結果見圖2。由圖2 可知:當水合氧化鋯吸附劑用量從0.5 g/L 增至2.0 g/L 時,溶液中氟質量濃度迅速由34.92 mg/L 降至8.54 mg/L,吸附劑的平衡吸附容量由34.92 mg/g 降至20.73 mg/g。繼續增加吸附劑用量,氟質量濃度下降趨勢逐漸趨于平緩,當吸附劑用量增至8 g/L 時,殘留氟質量濃度降至2.92 mg/L,吸附劑吸附容量降至5.89 mg/g。從吸附劑用量經濟因素考慮,確定水合氧化鋯吸附劑用量為2 g/L。
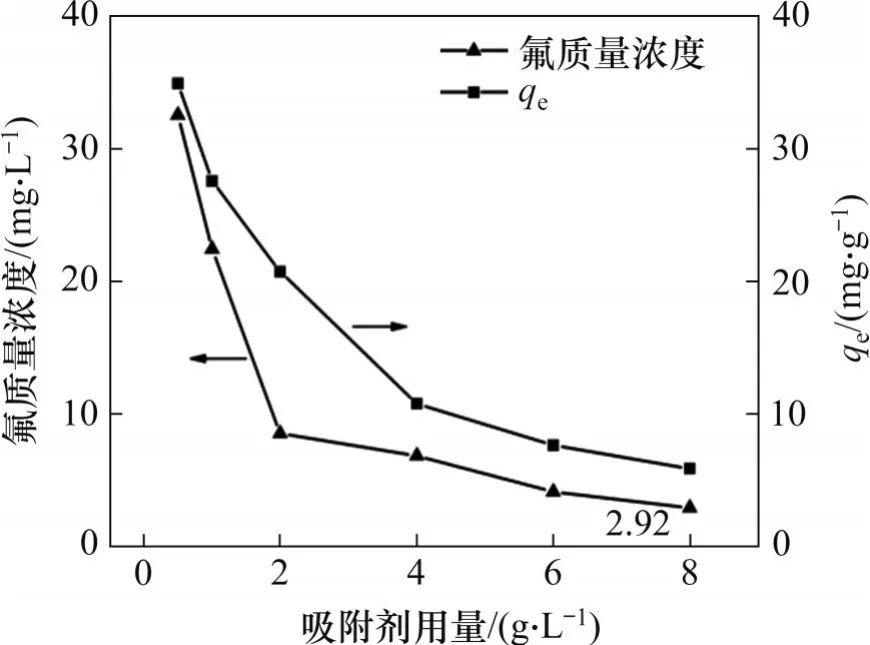
圖2 吸附劑用量對吸附效果的影響Fig.2 Effect of dose of adsorbent on adsorption efficiency
2.3 吸附動力學
動力學參數是衡量吸附劑吸附性能的一個重要指標。為考察吸附時間對水合氧化鋯吸附氟離子效果的影響,在pH=4.0,吸附劑用量為2 g/L,起始氟質量濃度為50 mg/L的條件下進行吸附動力學實驗,結果見圖3。
由圖3 可知:在前3 h 內,吸附劑表現出對氟離子的快速吸附,這是因為吸附劑表面尚存在較多吸附活性位點,隨吸附時間延長,活性位點逐漸被占據,表現為從3~5 h內吸附速率呈現下降趨勢,當吸附8 h時,體系達到吸附平衡,此時吸附劑的平衡吸附容量為20.73 mg/g,溶液中氟質量濃度下降至8.54 mg/L。進一步延長吸附時間,溶液中氟質量濃度基本保持不變。
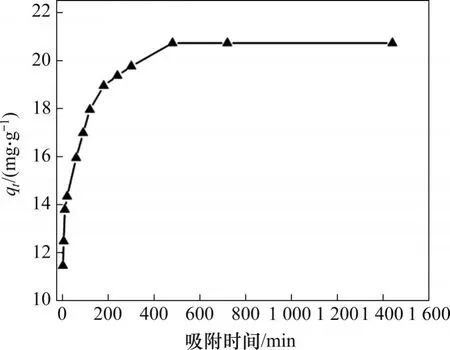
圖3 吸附時間對吸附效果的影響Fig.3 Effect of adsorption time on adsorption efficiency
為進一步描述硫酸錳溶液中的氟離子在水合氧化鋯吸附劑上的吸附過程,分別采用擬一級動力學模型和擬二級動力學模型對實驗結果進行擬合,2種模型的線性表達式分別為:
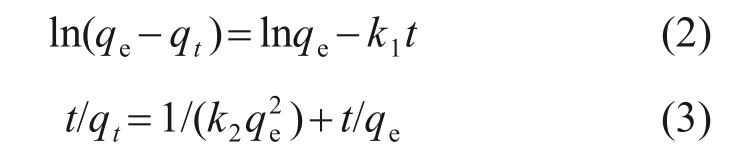
式中:k1和k2分別為擬一級動力學模型和擬二級動力學模型的吸附速率常數;t為吸附時間;qt為t時刻時吸附劑的吸附容量。動力學模型擬合結果見表1。
由表1 可見:擬二級動力學模型擬合度R2>0.99,比擬一級動力學模型的擬合度高,且由擬二級動力學模型擬合計算出吸附劑吸附容量為20.73 mg/g,與實驗值保持一致,進一步證明了硫酸錳溶液中的氟離子在水合氧化鋯上吸附可用擬二級動力學模型描述。擬二級動力學模型認為吸附速率由被吸附物質與吸附劑之間發生的化學反應決定,以此推測,硫酸錳溶液中氟離子在水合氧化鋯上的吸附可能主要屬于化學吸附,氟離子與吸附劑之間可能形成了新的化學鍵[12]。

表1 動力學模型擬合參數Table 1 Dynamic model fitting parameters
2.4 吸附等溫線
吸附等溫線模型參數是評價吸附材料性能的另一重要指標。為同時考察溫度對吸附效果的影響,在pH=4.0,吸附劑用量為2 g/L,吸附時間為8 h的條件下,考察5,20和35 ℃溫度下的吸附等溫線,結果見圖4。
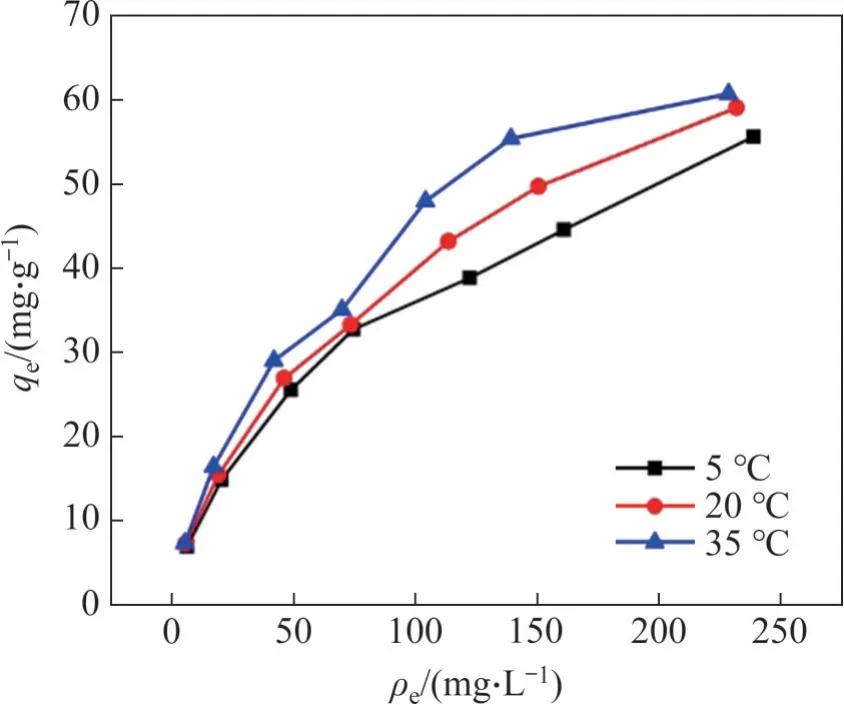
圖4 不同溫度下吸附等溫線Fig.4 Adsorption isotherms with different temperatures
由圖4可知:水合氧化鋯吸附劑吸附容量隨氟平衡質量濃度升高而增大;在35 ℃下,起始氟質量濃度350 mg/L,水合氧化鋯對氟離子的平衡吸附容量達到60.74 mg/L,這說明水合氧化鋯在高氟質量濃度下能達到較大吸附容量,即水合氧化鋯吸附劑同樣適用于吸附高氟硫酸錳溶液中氟離子;水合氧化鋯的平衡吸附容量明顯比硫酸鋁改性活性氧化鋁[9]的平衡吸附容量(1.96 mg/g)高。此外,溫度升高有利于氟離子在水合氧化鋯上的吸附,水合氧化鋯的吸附容量增大。
采用Langmuir 和Freundlich 這2 種吸附等溫模型對實驗數據進行擬合,2 種模型線性表達式分別為:
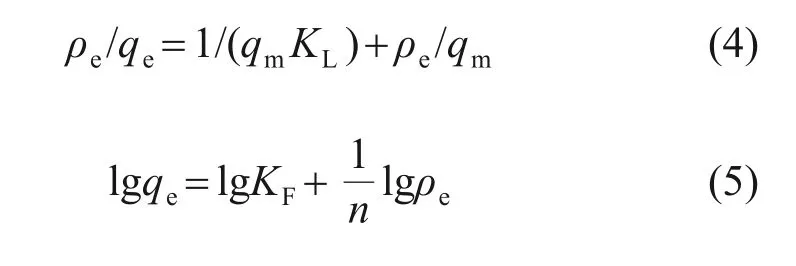
式中:KL和KF分別為Langmuir 吸附平衡常數和Freundlich吸附經驗常數;qm為Langmuir吸附等溫模型理論最大平衡吸附容量;n為與溫度有關的常數。
不同溫度下2 種模型擬合結果見表2。由表2可知:Freundlich 吸附等溫模型能較好地描述氟的吸附過程,氟在水合氧化鋯上的吸附更傾向于多層吸附[10]。3個溫度下水合氧化鋯對氟均顯示良好的吸附性能,1/n均在0.5左右,說明吸附較容易進行[13],吸附劑在室溫范圍內都能實際應用。隨溫度升高,Langmuir 吸附等溫模型擬合的qm和KL均變大,升溫有利于吸附,35 ℃時,水合氧化鋯對硫酸錳溶液中氟離子理論最大平衡吸附容量達77.64 mg/g。
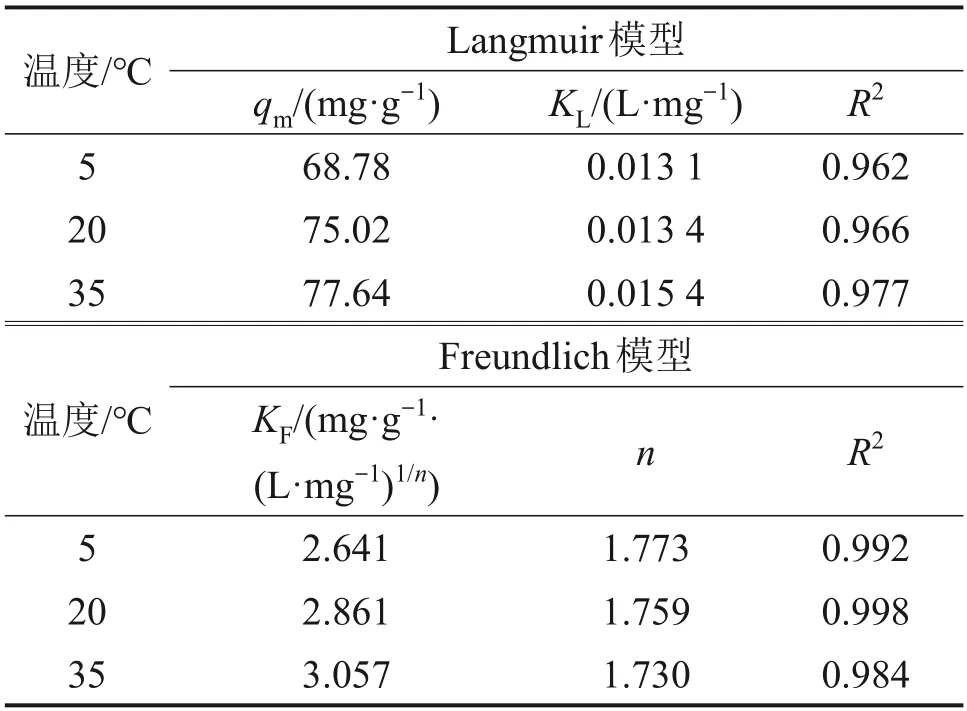
表2 Langmuir和Freundlich吸附等溫模型擬合參數Table 2 Fitting parameters of Langmuir and Freundlich adsorption isotherm model
2.5 吸附機理研究
圖5所示為水合氧化鋯吸附前后的SEM照片,吸附前后水合氧化鋯均呈不規則顆粒狀,吸附后吸附劑表面形貌并未發生明顯變化。水合氧化鋯吸附前后的FT-IR 圖如圖6 所示。由圖6 可見:在3 419 cm-1和1 615 cm-1的峰分別歸屬吸附劑表面吸附水的—OH 拉伸及彎曲振動吸收峰[14-15]。1 540 cm-1與1 358 cm-1峰歸屬于Zr—OH彎曲振動吸收峰[16],在吸附氟后,這2 個峰峰強度明顯減弱,表明吸附劑中上的羥基參與了吸附過程。在1 409 cm-1出現了新峰,可能是由于形成了Zr—F鍵[17]。此外,吸附后在1 132,1 063 和991 cm-1出現的峰屬SO42-特征峰[18],表明吸附劑對溶液中硫酸根存在一定吸附作用。

圖5 水合氧化鋯吸附前后SEM照片Fig.5 SEM images of hydrous zirconium oxide before and after adsorption
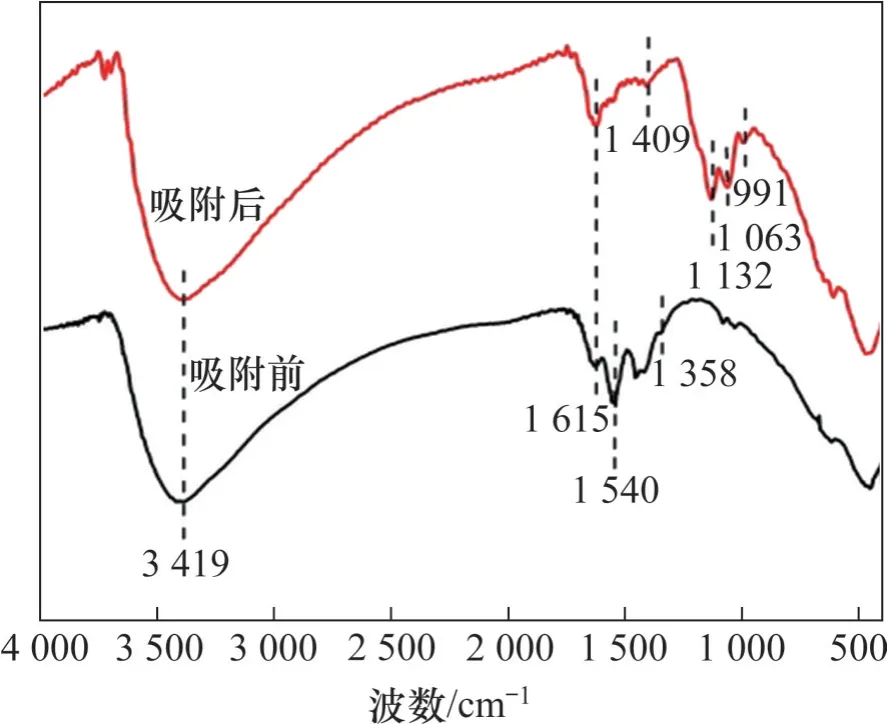
圖6 水合氧化鋯吸附前后FT-IR圖Fig.6 FT-IR images of hydrous zirconium oxide before and after adsorption
圖7 所示為水合氧化鋯吸附前后XPS 全譜圖,由圖7可知:吸附后出現了F 1s新峰,證明溶液中氟離子成功吸附在水合氧化鋯上。氟的能譜圖如圖8所示。由圖8可見:F 1s峰位置位于684.91 eV處,介于NaF 的684.5 eV 與ZrF4的685.3 eV 之間,說明鋯與氟之間形成了化學鍵[19]。吸附前后吸附劑各原子數百分數見表3,由表3 可知:吸附氟離子后,吸附劑上夾帶的錳原子數僅占吸附劑總原子數的0.38%,這說明水合氧化鋯除氟后錳損失極低,這是由于水合氧化鋯吸附劑吸附容量大,用量少,從而減少了錳的夾帶[20]。ICP檢測吸附后硫酸錳溶液中鋯殘留僅為0.006 mg/L,水合氧化鋯在除氟過程中基本不溶出。可見,水合氧化鋯具有除氟效率高、錳損失少、不引入雜質的優點,適合用于電池級硫酸錳溶液中除氟。
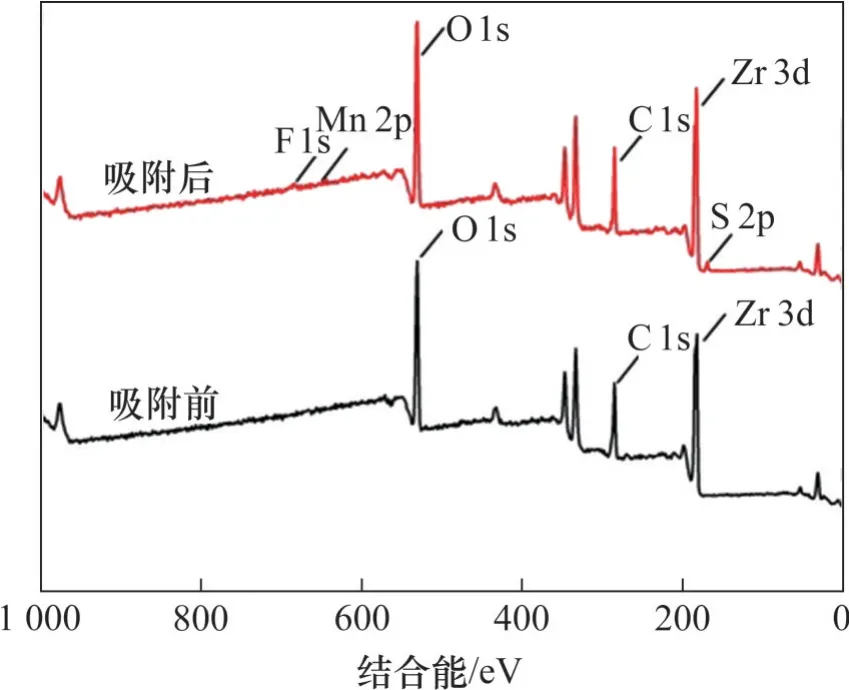
圖7 水合氧化鋯吸附前后XPS全譜圖Fig.7 XPS survey spectra of hydrous zirconium oxide before and after adsorption
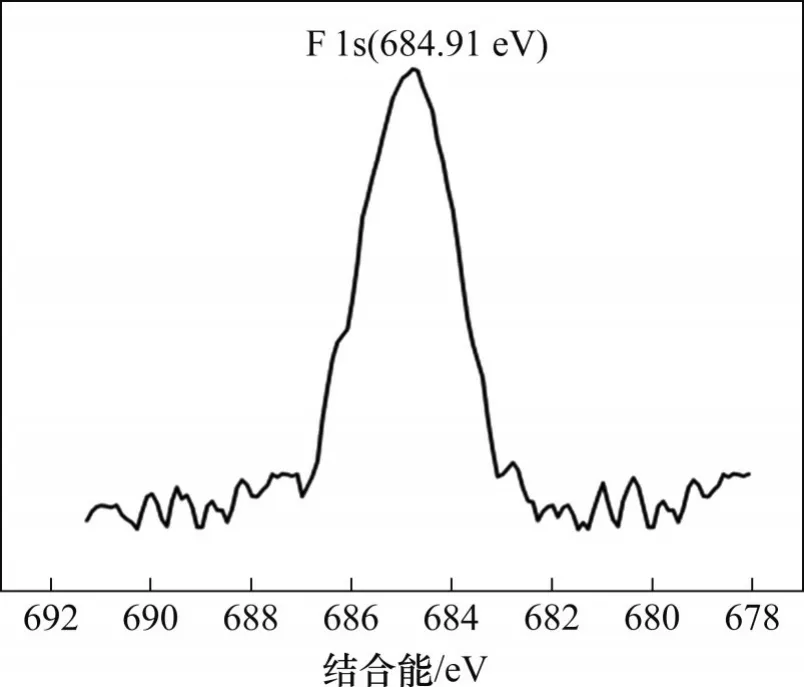
圖8 水合氧化鋯吸附后F 1s能譜圖Fig.8 F 1s spectra of hydrous zirconium oxide after adsorption
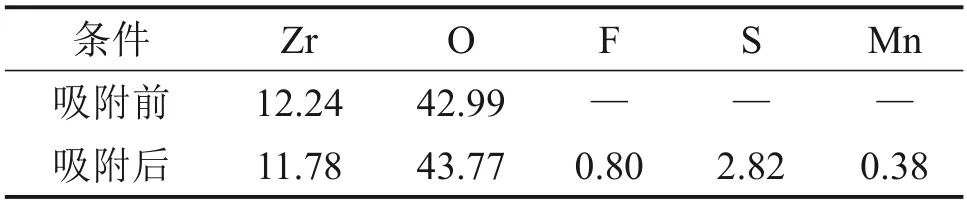
表3 吸附劑吸附前后各原子數百分數Table 3 Atomic quantity percentage of adsorbent before and after adsorption %
吸附前后Zr能譜圖如圖9所示,由圖9可以看出:吸附前,Zr 3d3/2和Zr 3d5/2峰分別位于184.35 eV和182.03 eV 處,吸附氟離子后,Zr 3d3/2和Zr 3d5/2峰位置向高結合能方向移動,移至184.82 eV 和182.44 eV,分別增加了0.47 eV和0.41 eV,這證實了吸附劑上羥基被氟取代形成了Zr—F鍵,這是由于鋯與氟成鍵引起鋯電子密度下降,進而提高了Zr 3d3/2和Zr 3d5/2的結合能[21-22]。O 1s 能譜圖如圖10 所示。由圖10 可見:吸附前分為3 個峰,分別在533.07,531.42 和530.12 eV 處,依次歸屬于吸附劑表面吸附水(H2O)、吸附劑表面羥基(Zr—OH)、金屬氧化物(Zr—O),吸附氟離子后,Zr—OH 的峰面積占比由吸附前的57.75% 下降至36.71%,說明吸附劑中的羥基參與了吸附過程;吸附后出現在532.30 eV 處的新峰歸屬于S—O 鍵的特征峰[23]。結合FT-IR譜圖分析可知,氟離子通過與水合氧化鋯中的羥基發生離子交換而去除。
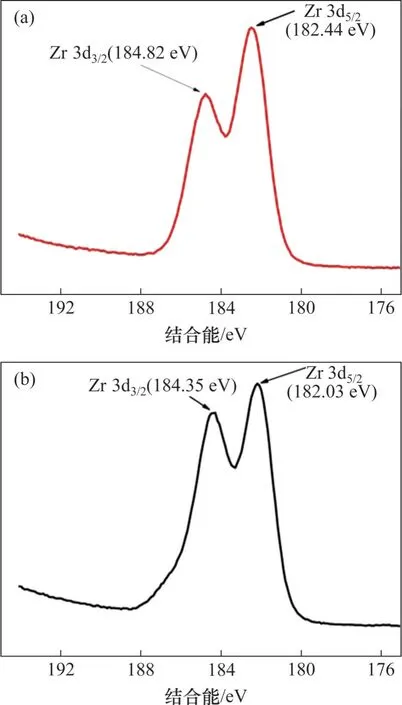
圖9 水合氧化鋯吸附前后Zr 3d能譜圖Fig.9 Zr 3d spectra of hydrous zirconium oxide before and after adsorption
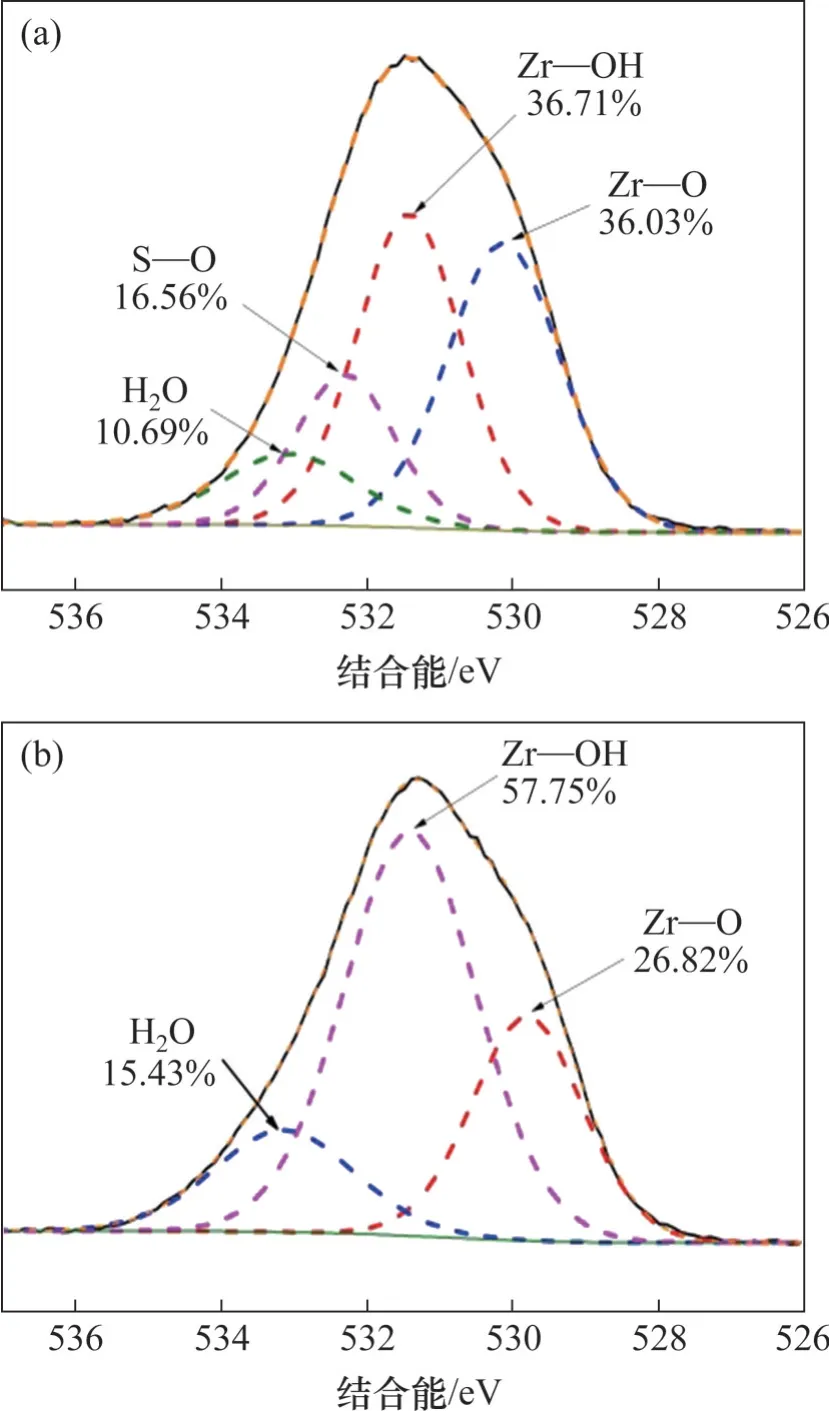
圖10 水合氧化鋯吸附前后O 1s能譜圖Fig.10 O 1s spectra of hydrous zirconium oxide before and after adsorption
3 結論
1) 在pH 為4.0、吸附劑用量為2 g/L、吸附溫度為20 ℃、吸附時間為8 h的條件下,硫酸錳溶液中氟質量濃度可由50 mg/L降至8.54 mg/L。
2) 硫酸錳溶液中氟離子在水合氧化鋯上吸附符合擬二級動力學模型和Freundlich 吸附等溫模型,升溫有利于吸附,35 ℃下水合氧化鋯對氟離子理論最大平衡吸附容量達77.64 mg/g。
3) 氟離子通過與吸附劑中的羥基發生離子交換而被去除。
4) 水合氧化鋯除氟后溶液中的鋯殘留量與錳損失均極低,不引進新雜質,可保證產品純度達到電池級硫酸錳要求,水合氧化鋯適用于電池級硫酸錳溶液中氟的去除。
猜你喜歡
中學生數理化·中考版(2022年10期)2022-11-10 09:37:42
中學生數理化·八年級物理人教版(2022年12期)2022-02-14 07:08:42
中學生數理化·八年級物理人教版(2021年12期)2021-12-31 03:23:08
中學生數理化·中考版(2020年10期)2020-11-27 01:59:48
中國生殖健康(2019年2期)2019-08-23 08:12:08
石油化工建設(2018年6期)2018-04-22 03:16:54
產品可靠性報告(2017年7期)2017-09-05 09:49:12
中學生數理化·八年級物理人教版(2017年12期)2017-04-18 12:59:38
汽車觀察(2016年3期)2016-02-28 13:16:26
民生周刊(2014年7期)2014-03-28 01:30:54
