極長鏈酰基輔酶A脫氫酶缺乏癥誤診為肝糖原累積癥1例*
2023-03-02 09:03:14朱文芳孫克偉
中西醫結合肝病雜志 2023年2期
關鍵詞:肝功能
袁 維 朱文芳 陳 斌 杜 珊 孫克偉
湖南中醫藥大學第一附屬醫院肝病科 (湖南 長沙,410007)
極長鏈酰基輔酶A脫氫酶缺乏癥(VLCADD,OMIM 201475)是一種是由于極長鏈酰基輔酶A脫氫酶(VLCAD)的編碼基因ACADVL(OMIM609575)先天缺陷導致長鏈脂肪酸氧化障礙的一種常染色體隱性遺傳病,是發病率居第二位的先天性脂肪酸氧化障礙性疾病[1]。VLCAD是線粒體內脂肪酸β氧化中的第一步關鍵酶,催化含14~18個碳的脂酰基輔酶A脫氫[2],其缺陷導致脂肪酸β氧化過程受阻、供能障礙及長鏈酰基肉堿蓄積,從而對心肌、肝臟、骨骼肌等產生毒性作用。由于VLCADD發病率低,臨床表現存在異質性,常規實驗室檢查診斷較困難,常容易誤診及漏診。湖南中醫藥大學第一附屬醫院2021年8月收治1例VLCADD患者,現將其臨床資料及相關文獻回顧性總結如下。
1 病例資料
2015年7月患者因“口干、多飲、多尿3年,腹痛、頭痛半月余”于當地醫院就診,查肝功能:AST 74.7 U/L,ALT 59.6 U/L;腹部B超:肝大,右中腹腔腸系膜多發淋巴結增大,上腹腔腸氣增多;腦電圖輕度異常;TG 1.9 mmo1/L,經對癥支持治療后患者自覺癥狀緩解,復查肝功能正常,腦電圖未見異常,出院診斷:①Ⅰ型糖尿病、糖尿病腎病;②腸系膜淋巴結炎;③癲癇;④膽汁返流性胃炎。
2018年10月患者再次前往當地復查肝功能:AST 104.8 U/L,ALT 74.3 U/L,TBA 14.1 μmo1/L;腎功能:CREA 36.5 μmo1/L;血脂:TG 2.01 mmol/L,HDL-C 1.00 mmo1/L;空腹C肽0.06 mg/ml;病毒性肝炎全套、巨細胞病毒DNA定量、EB病毒DNA、銅藍蛋白(-);腹部彩超:①肝稍大(右肋緣下36 mm);②右中腹腔腸系膜多發淋巴結增大。經對癥支持治療患者自覺癥狀好轉,肝功能復查正常出院,出院后患者間斷服用肌苷片護肝,未定期復查肝功能。
2019年患者因“發熱、腹瀉、血糖升高”于當地醫院就診,住院治療時完善檢查提示:肝大,肝功能異常,行護肝、降糖等治療后病情好轉出院(具體不詳),出院后仍未定期復查肝功能。
2021年7月8日患者因血糖控制不佳在當地醫院住院治療,住院期間查肝功能:AST 319.00 U/L,ALT 361.60 U/L,余不詳;腎功能:UA 488.00 μmol/L;心肌酶:CK-MB 58.00 U/L,LDH 370.40 U/L;電解質:Na 133.40 mmol/L;肝膽胰脾彩超:左肝稍大;經護肝降酶等對癥支持治療后復查肝功能:AST 85.80 U/L,ALT 145.00 U/L,TP 59.00 g/L,ALP 202.20 U/L,γ-GGT 137.40 U/L。因患者反復肝大、肝功能異常原因不明確,為求進一步診治,于2021年8月4日前往我院門診就診,門診以“肝功能異常查因:肝糖原累積癥?”收住入院。入院癥見:患者神清,精神尚可,食欲尚可,睡眠差,二便正常,體重未減輕。
既往史:9年前發現“1型糖尿病”,使用三餐前重組賴脯胰島素15 IU+睡前重組甘精胰島素15 IU控制血糖,自訴血糖控制不佳,運動或饑餓后時有低血糖發生(具體不詳);2019年因外傷導致顱腦損傷(具體不詳);否認手術史;否認輸血史。否認藥、食過敏史,否認其他接觸物過敏史。否認傳染病史。
出生史及家族史:父母體健,非近親結婚,已離異,否認家族中類似疾病史。
入院查體:四測正常,營養欠佳,形體瘦小,慢性肝病面容,皮膚黝黑,皮膚及鞏膜無黃染,有肝掌,顏面及前胸未見蜘蛛痣。雙肺呼吸音清,未聞及干、濕啰音,未聞及胸膜摩擦音。心率:99次/min,律齊,各瓣膜區未聞及病理性雜音,未聞及心包摩擦音。肝上界位于右側鎖骨中線第5肋間,肝區叩擊痛(+),肝臟肋下3 cm可觸及,質中等,表面光滑,脾臟肋下未觸及。腹部平坦,腹壁靜脈隱現,腹軟,無壓痛及反跳痛,無腹肌緊張,未見胃腸型及蠕動波,Murphy′s征(-),無移動性濁音,腸鳴音正常,4次/min,腎區叩擊痛(-)。雙下肢無水腫。
住院期間完善相關檢查,血常規、大便常規未見異常;尿常規+沉渣:GLU(++++),余未見異常;凝血功能:PT 10.40 s↓,PT-INR 0.84↓;肝功能:ALT 85.60 IU/L↑,AST 82.90 IU/L↑;ALP 394.00 U/L↑,γ-GGT 67.00 U/L↑;腎功能:UA 542.00 μmol/L↑;體液免疫:IgA 2.58 g/L↑,余(-);心肌酶:CK-MB 30.60 IU/L↑,余正常;PCT 0.081 ng/ml↑;C肽0.02 ng/ml↓;餐后2 h C肽0.01 ng/ml;糖尿病自身抗體譜:谷氨酸脫羧酶抗體(GAD)>2 000 U/ml↑,胰島素自身抗體(IAA)1.48 U/ml↑;乳酸6.09 mmol/L↑;甲狀腺功能全套:甲狀腺過氧化酶抗體145.09 IU/ml↑,余正常;24 h尿銅13.7 μg;血清銅963.8 μg/L;血氨、B2微球蛋白測定、視黃醇結合蛋白、酮體、血清胱抑素測定、白介素6、CRP、電解質、淀粉酶、銅藍蛋白、甲胎蛋白未見異常;病毒性肝炎全套、HBV DNA、自免肝全套、抗ENA抗體全套、輸血前檢查、巨細胞病毒IgM抗體、葡萄糖-6-磷酸脫氫酶(G-6-PD)缺陷篩查陰性;心電圖、胸片未見明顯異常;腹部彩超:肝臟體積增大(肝右葉最大斜徑131 mm,肝左葉前后徑95 mm,左葉上下徑68 mm),肝實質光點增粗,請結合臨床;脾稍大(厚約36 mm,長約117 mm);余未見異常。雙側睪丸、附睪彩超:雙側睪丸微石癥;右側附睪頭多發囊腫;余未見異常;腎上腺彩超正常;左手正位(含左腕、骨齡片):左腕可見8枚骨化中心,第一掌骨近端骨骺及第二至第五指間骨骺出現,橈骨及尺骨遠端骨骺出現。腦垂體MRI:垂體上緣稍凹陷,考慮鞍上池部分腦脊液稍下限所致,余未見異常。肝臟彈性檢測:肝臟硬度8.6 kPa,脂肪衰減219 db/m。青少年男性患者,反復肝功能異常,高尿酸血癥、高脂血癥、血糖異常,排除了病毒性肝炎、自身免疫性肝病、非酒精性脂肪性肝病、藥物性肝病等常見肝病,結合患者病史,需考慮遺傳代謝性肝病,重點考慮肝糖原累積癥,故予以完善肝臟病理、遺傳病全外顯子組測序。
特殊檢查:①肝臟病理:光鏡下肝穿刺組織形態考慮為肝糖原累積癥,PAS染色(+),鐵染色(-)、網狀纖維染色示肝小葉結構保存(圖1);電鏡下個別肝細胞線粒體增大伴結晶形成,少部分肝細胞糖原顆粒輕度增加,少許小脂滴和少量淤膽性色素顆粒,余未見特殊超微病理改變(圖2);②基因分析:經醫院醫學倫理委員會批準以及在家屬知情同意的前提下,抽取患者、患兒母親外周靜脈血送檢基因公司行遺傳病全外顯子組測序,結果提示ACADVL基因編碼區發生的移碼變異,患兒ACADVL 等位基因上有1個來源于母親的移碼變異:NM_000018.4(c.266delC∶p.Pro89Hisfs*28)(圖3)。其他遺傳病基因未檢測到。因患者肝臟病理電鏡檢查未見糖原累積依據,基因檢測未見糖原累積癥相關基因改變,因此本例最終考慮診斷為VLCADD。
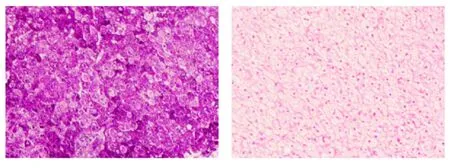
圖1 患者肝臟病理(光鏡)
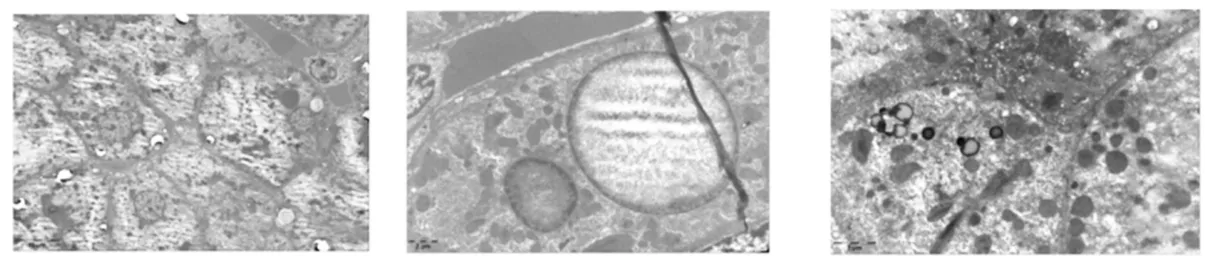
圖2 患者肝臟病理(電鏡)
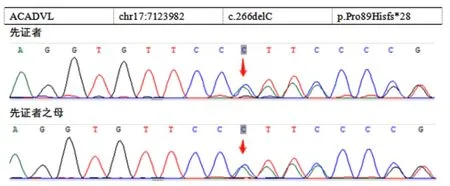
注:c.266delC:p.Pro89Hisfs*28,編碼區第266號核苷酸胞嘧啶缺失,導致所編碼蛋白質自第89號脯氨酸開始發生編碼紊亂,為移碼變異。
治療經過:經予以護肝、降血糖、補充維生素、限制長鏈甘油三酯(LCT)攝入并補充中鏈甘油三酯(MCT)、控制血糖等對癥支持治療后,患者自覺癥狀好轉,隨訪至今,患者一般情況良好。
2 討論
VLCADD最早于1993年由Bertrand等[3]報道。VLCADD發病率低,在1∶3萬到1∶40萬之間。VLCADD致死率高,為嬰幼兒期潛在猝死性疾病之一,病情極易反復,嚴重威脅嬰幼兒的健康。目前VLCADD 已被列入新生兒遺傳代謝病首要篩查的病種中,以達到早篩查、早診斷和早治療的目的。
VLCADD 的臨床表現根據起病年齡和器官系統受累程度分心肌病型、肝型、肌病型3個類型[1]:①心肌病型:又稱新生兒型,主要在新生兒和嬰兒早期發病,是最常見的一種嚴重早發心臟和多臟器衰竭型。此型發病兇險,患兒病死率高,表現為瑞氏綜合征、新生兒猝死、肥厚型和擴張型心肌病、心包積液、心律失常、肌無力、肝腫大和間歇性低血糖,肌酸激酶水平升高;心肌肥厚和心律失常可致死。②肝型:又稱為嬰兒型,嬰兒后期或兒童期發病,病情較輕,為肝臟和低酮性低血糖型,表現為肝大和肝功能異常,空腹耐力下降和急性低酮性低血糖,不伴心肌損害和心肌肥厚,但未經及時診斷和治療也會有生命危險。③肌病型:又稱為晚發型,多在青少年甚至成年期發病,癥狀輕,主要表現為運動、感染或饑餓后的橫紋肌溶解和肌紅蛋白尿,其至可發生腎功能衰竭,可伴有肌無力、肌肉痛性痙攣或肌痛。急性失代償導致死亡而沒有明顯的前驅癥狀,類似嬰兒猝死綜合征(SIDS),也曾在晚期診斷患者中描述過[4]。
VLCADD的臨床表現存在明顯的異質性。本例患者6年前開始出現肝大和反復肝功能異常,時有運動或饑餓后低血糖發作,符合肝病型的特點。該患者同時合并高乳酸血癥及高尿酸血癥,多次發病,其中2次因為炎癥及感染后發現肝功能異常,多次運動及饑餓后出現低血糖,予對癥治療后好轉;肝病型病情比心肌病型輕,但如若診治不及時,同樣也可能危及生命。
VLCAD診斷包括串聯質譜血酯酰肉堿譜分析、尿氣相質譜有機酸分析、成纖維細胞脂肪酸β-氧化流量分析、VLCAD酶活性和ACADVL基因分析[5,6]。串聯質譜血酰基肉堿譜中肉豆蔻烯酰基肉堿(C14∶1)的升高是診斷VLCADD 最重要的生化代謝指標[6]。但在非應激期間或已經進食、靜脈葡萄糖輸注的患者及臨床表現較輕的患者中可能出現假陰性。美國NBS研究基于標準串聯質譜方法預測酰基肉堿分析的VLCADD陽性預測值僅為20%~30%[5,7]。尿氣相質譜有機酸分析可發現二羧酸尿癥[5],但輕癥患者或伴有橫紋肌溶解患者可無二羧酸尿癥。可對患者的皮膚成纖維細胞、外周血淋巴細胞、心肌和骨骼肌細胞或組織進行酶學分析測定VLCAD活性明確診斷。但在培養的皮膚成纖維細胞或淋巴細胞中測定中發現β-氧化流量僅在60%的病例中降低,表明這些方法檢測VLCAD缺陷的敏感性有限[8]。病理檢查可見肝臟脂肪變性,心肌、骨骼肌脂質沉積。
ACADVL基因(OMIM 609575)突變分析檢出2個等位基因致病突變是確診VLCADD的金標準[3]。ACADVL基因位于染色體17p13.1,長約5.4 kb,含20個外顯子,編碼655個氨基酸,目前已有381個突變被HGMD數據庫收錄(http://www.hgmd.cf.ac.uk/ac/index.php)。既往研究均提示ACADVL基因的突變譜高度異質,暫無明確熱點突變,且新突變、疑似致病或臨床意義不明的突變位點較多,其中錯義突變為主要的突變類型,約占60.1%,缺失突變約占16.5%,剪接突變約占12.1%。一般基因檢測僅有80%病例可明確診斷。
本研究全外顯子組測序結果提示患者ACADVL基因僅其中一個等位基因上發生了移碼變異:NM_000018.4(c.266delC∶p.Pro89Hisfs*28),遺傳自母親,未檢測到其他疾病相關基因改變。毋庸置疑,基因檢測在只發現1個致病突變或新的臨床意義未明突變時,診斷將會變得困難。Laforêt等[8]檢測了13例來自10個不同家族的VLCAD缺陷成人患者,其中2例患者只檢測到一個雜合狀態的突變,研究者認為在檢測區域之外可能存在對ACADVL基因表達至關重要的其他突變,例如內含子深部的變異或基因表達調控序列。Marcus等[9]對693例VLCADD新生兒篩查為陽性者進行了分子檢測,發現57%的個體存在單一致病變異或臨床意義未明的變異,未能提供另一個等位基因存在第二個致病變異的證據。作者認為這種常見的攜帶者狀態可能是由于Sanger序列分析遺漏了第二個致病基因。另外,影響深層內含子或啟動子序列的變異也可能被研究者提供的Sanger分析所遺漏。該文獻報道在VLCADD患者中檢出本例患者類似變異[9],提示該變異所致氨基酸序列缺失部分對蛋白質功能有重要作用。該變異在大規模人群頻率gnom AD數據庫報道了1例雜合個體,未見純合個體報道,總體人群頻率為0.000 003 977。根據現有證據,結合患者臨床表現,考慮該患者可明確診斷為VLCADD。
VLCADD治療策略包括通過提供足夠的能量和營養攝入來預防分解代謝發作,避免過度禁食(特別是在生病期間),避免脫水、高脂飲食、感染、勞累和心肌刺激等情況,限制LCT來減少異常脂肪酸代謝產物的產生,給予高碳水化合物和低脂飲食,但需保證必需脂肪酸的攝入,補充MCT,同時需提供足夠的蛋白質,對癥處理及預防并發癥。最佳的VLCAD管理需要持續評估患者的臨床表現及營養狀況。特殊營養補充的需求(即維生素A、D、E、DHA和其他)也被評估[1]。目前尚不清楚在VLCADD患者中使用肉堿是否安全以及預防肉堿缺乏的益處是否超過長鏈酰基肉堿累積的潛在心臟風險。Watanabe等[10]報道了2例VLCADD患者在補充左旋肉堿后發生橫紋肌溶解。建議僅在必要時口服肉堿以防止血漿中肉堿缺乏。苯扎貝特雖被證實能增加VLCAD缺陷患者細胞中棕櫚酸酯的氧化作用,但體內有效性仍存在爭議[11,12]。隨著對脂肪酸氧化酶調控機制研究的深入,通過翻譯后修飾的調節來促進酶活性已經成為重要的新的治療靶點,尤其在VLCADD上取得了更多的進展,無論是在小鼠模型還是VLCADD患者的纖維母細胞中[13]。雖然目前正在研究其他新型治療藥物(奇數鏈脂肪酸三庚酸可進一步改善運動耐力和耐受性以及心臟功能)[14-16],但仍存在哪些患者需要治療以及多種ACADVL基因變異的臨床意義的問題[17,18]。基因治療仍是實現黃金治療所有遺傳疾病的努力方向。其他治療包括通過葡萄糖液體水合和堿化尿液來治療急性橫紋肌溶解癥,以保護腎功能及預防繼發于肌紅蛋白尿癥的急性腎衰竭,糾正低血糖,及時退熱等(高溫可使VLCAD活性降低)。定期監測包括心臟狀況在內的年度體檢、一年或兩年一次的超聲心動圖檢查以及生長和營養狀況的年度評估,運動前MCT似乎可以預防骨骼肌疾病[19,20]。
肝糖原累積癥是一種先天性糖原代謝紊亂性疾病,常染色體隱性遺傳病,是由于糖原合成或分解過程中某些酶的缺陷或結構異常,導致機體內各種組織細胞內糖原異常增多的一類疾病,臨床表現為明顯低血糖、高膽固醇、高甘油三酯血癥、高尿酸血癥。本例患者因肝功能異常、肝大、血糖異常、高尿酸血癥、高脂血癥等表現就診,臨床診斷最初考慮肝糖原累積癥可能性大,肝臟病理光鏡檢查也支持糖原累積綜合征的診斷,但該患者肝臟電鏡檢查未提示肝糖原累積癥,且全基因外顯子組測序未發現糖原累積癥相關的基因改變,結合患者的臨床表現及外顯子測序本例最終明確診斷為VLCADD(肝型)。因此,在臨床工作中若遇到不明原因的肝大、肝功能異常、酸中毒、高乳酸血癥、高脂血癥、心肌酶異常等情況的患者,要盡早進行遺傳代謝病相關篩查,對于原因仍然不能明確的,建議盡早行基因檢查,全外顯子組測序能提高診斷率,同時可提供遺傳咨詢,指導優生優育。
猜你喜歡
肝博士(2024年1期)2024-03-12 08:38:08
傳染病信息(2022年6期)2023-01-12 08:58:58
肝博士(2022年3期)2022-06-30 02:48:58
昆明醫科大學學報(2021年3期)2021-07-22 07:39:56
中外醫療(2016年15期)2016-12-01 04:25:40
中國衛生標準管理(2015年1期)2016-01-14 03:41:20
海軍醫學雜志(2015年2期)2015-02-27 13:47:43
癌變·畸變·突變(2015年4期)2015-02-27 06:15:18
中國藥業(2014年12期)2014-06-06 02:17:26
中國藥業(2014年19期)2014-05-17 03:12:13
