濁點(diǎn)萃取—異辛烷反萃取—?dú)庀嗌V質(zhì)譜法測(cè)定茶飲料中8種農(nóng)藥殘留
2023-03-22 13:02:34孔令燦孟元華
食品與機(jī)械 2023年1期
關(guān)鍵詞:方法
謝 潔 周 閏 孔令燦 孟元華 王 宇
(1.無錫市疾病預(yù)防控制中心,江蘇 無錫 214023;2.南京醫(yī)科大學(xué),江蘇 南京 211166)
隨著生活節(jié)奏的不斷加快,人們對(duì)茶產(chǎn)品的要求逐漸向方便化趨勢(shì)發(fā)展,茶,正以時(shí)尚化、便捷化的方式進(jìn)入人們的生活。隨著茶飲料的大量普及,相應(yīng)的安全問題也受到社會(huì)的大量關(guān)注,如農(nóng)殘檢測(cè)[1]。在茶飲料中,農(nóng)殘含量相對(duì)較低,按照目前傳統(tǒng)的檢測(cè)方法,在檢測(cè)中需要用到大量的有機(jī)溶劑,更需要花費(fèi)大量時(shí)間在濃縮提取過程上,操作繁瑣,費(fèi)時(shí)費(fèi)力。
國(guó)內(nèi)外對(duì)于農(nóng)藥殘留的分析報(bào)道中,儀器方法主要有:免疫分析技術(shù)[2]、氣相色譜[3]、毛細(xì)管電泳技術(shù)[4]、氣相色譜—質(zhì)譜聯(lián)用[5-6]、液相色譜—質(zhì)譜聯(lián)用[7]、超高效液相色譜—質(zhì)譜聯(lián)用[8-9]等。其中,樣品前處理技術(shù)多為分散固相萃取[10-11]、分散液液微萃取[12]、固相萃取柱凈化[6]、QuEChERS凈化[13-14]等,在操作過程中均需使用大量有機(jī)溶劑,耗時(shí)長(zhǎng),操作步驟繁瑣,且需要復(fù)雜的凈化步驟。濁點(diǎn)萃取法(cloud point extraction,CPE)能在一定程度上避免上述問題,研究擬以表面活性劑水溶液作為萃取劑,通過表面活性劑的增溶性和濁點(diǎn)分相來對(duì)目標(biāo)物進(jìn)行分離和富集,表面活性劑中的目標(biāo)物則通過微量的有機(jī)溶劑萃取出來,該方法不但能提高富集倍數(shù),同步完成待測(cè)組分的提取和凈化,也能有效避免表面活性劑的高黏度對(duì)檢測(cè)儀器產(chǎn)生的影響。該技術(shù)目前已被廣泛應(yīng)用于重金屬分析[15]、食品工業(yè)[16-17]、金屬離子形態(tài)分析[18-19]、環(huán)境污染物檢測(cè)[20-21]、環(huán)境有機(jī)分析[22]等領(lǐng)域,但是關(guān)于濁點(diǎn)萃取反萃取法同時(shí)富集分離茶飲料中多種農(nóng)藥的研究尚未見報(bào)道。
研究擬以非離子表面活性劑PEG 4000作為萃取劑從茶葉飲料中提取和預(yù)濃縮8種農(nóng)殘,再?gòu)墨@得的富含表面活性劑相中反萃取到異辛烷中。從而建立從茶飲料中同時(shí)提取檢測(cè)多種農(nóng)藥殘留的濁點(diǎn)萃取方法,以期為茶飲料中擬除蟲菊酯和有機(jī)氯類農(nóng)藥殘留的檢測(cè)提供參考。
1 材料與方法
1.1 材料
1.1.1 主要儀器
氣相色譜/質(zhì)譜儀:7890B-7000C型,美國(guó)Agilent公司;
超純水系統(tǒng):Milli-Q型,美國(guó)Millipore公司;
高速冷凍離心機(jī):3-30K型,美國(guó)Sigma公司;
電子天平:XS105DU型,美國(guó)Mettler Toledo公司;
超聲儀:SCQ-10018型,上海聲彥超聲波儀器公司;
渦旋混合器:G560型,美國(guó)Scientific Industries公司。
1.1.2 試劑與材料
艾氏劑(CAS:309-00-2)、狄氏劑(CAS:60-57-1)、六氯苯(CAS:118-74-1)、七氯(CAS:76-44-8)、氯氰菊酯(CAS:52315-07-8)、甲氰菊酯(CAS:64257-84-7)、溴氰菊酯(CAS:52918-63-5)、氰戊菊酯(CAS:51630-58-1):純度≥99.5%,德國(guó)Dr.Ehrenstorfer公司;
正己烷:色譜純,德國(guó)默克公司;
異辛烷:色譜純,上海阿拉丁生化科技股份有限公司;
石油醚:色譜純,美國(guó)TEDIA公司;
茶飲料6種:市售;
PEG 4000:分析純,韓國(guó)樂天公司;
Tween-20:分析純,德國(guó)默克公司;
Triton X-100:分析純,美國(guó)Sigma公司;
氯化鈉、無水硫酸鈉:分析純,國(guó)藥集團(tuán)化學(xué)試劑有限公司;
試驗(yàn)用水:符合GB/T 6682—2008《分析實(shí)驗(yàn)室用水規(guī)格和試驗(yàn)方法》規(guī)定的一級(jí)水。
1.2 方法
1.2.1 農(nóng)藥標(biāo)準(zhǔn)溶液的配制 分別取適量8種農(nóng)藥標(biāo)準(zhǔn)品,用正己烷配制成質(zhì)量濃度為5 mg/L的4種有機(jī)氯類農(nóng)藥和質(zhì)量濃度為10 mg/L的4種擬除蟲菊酯類農(nóng)藥的混合標(biāo)準(zhǔn)中間液,-20 ℃保存?zhèn)溆茫行跒?個(gè)月。再用異辛烷逐級(jí)稀釋,有機(jī)氯類農(nóng)藥逐級(jí)稀釋為2.0,1.0,0.5,0.1,0.05,0.01 mg/L的混合標(biāo)準(zhǔn)儲(chǔ)備液,擬除蟲菊酯類農(nóng)藥逐級(jí)稀釋為4.0,2.0,1.0,0.2,0.10,0.02 mg/L的混合標(biāo)準(zhǔn)儲(chǔ)備液,均保存于4 ℃,有效期為2~3周。
1.2.2 樣品前處理
(1)濁點(diǎn)萃取方法:采集市場(chǎng)上不同品牌的6個(gè)茶飲料樣品,準(zhǔn)確移取10.0 mL于25 mL刻度試管中,依次準(zhǔn)確加入300 g/L的PEG 4000溶液2.0 mL、無水硫酸鈉1.0 g,混合均勻后經(jīng)超聲輔助至全部溶解。將上述混合溶液于40 ℃水浴20 min。待其靜置分層后用長(zhǎng)細(xì)針頭的注射器吸出水相棄去。
(2)反萃取方法:向剩下的表面活性劑相中準(zhǔn)確加入異辛烷200 μL,漩渦混合1 min,待兩項(xiàng)分層后,移液槍吸取上層異辛烷相至自動(dòng)進(jìn)樣小瓶的內(nèi)襯管中,待測(cè)。
(3)加標(biāo)回收方法:取空白茶飲料18份,分別添加低、中、高3種不同濃度(六氯苯、七氯、艾氏劑、狄氏劑4種農(nóng)藥的低、中、高質(zhì)量濃度為0.01,0.10,1.0 μg/L,聯(lián)苯菊酯、甲氰菊酯、高效氯氟氰菊酯、氯氰菊酯4種農(nóng)藥的低、中、高質(zhì)量濃度為0.02,0.20,2.0 μg/L)的標(biāo)準(zhǔn)溶液進(jìn)行加標(biāo)回收試驗(yàn),每個(gè)加標(biāo)水平重復(fù)6次。
1.2.3 色譜條件 進(jìn)樣口溫度280 ℃;進(jìn)樣量1 μL,不分流進(jìn)樣;恒流模式,流速1 mL/min;Agilent HP-5 ms UI毛細(xì)管色譜柱(30 m×0.25 mm×0.25 μm);柱升溫程序:60 ℃保持2 min,以20 ℃/min升溫至120 ℃并保持1 min,以10 ℃/min升溫至250 ℃并保持3 min。
1.2.4 質(zhì)譜條件 在選擇性離子(SIM)模式下檢測(cè),采用電子轟擊源(EI+);電壓70 eV;傳輸線溫度150 ℃,離子源溫度230 ℃,四極桿溫度150 ℃,溶劑延遲時(shí)間10 min。
1.2.5 數(shù)據(jù)處理 采用安捷倫Mass Hunter workstation軟件進(jìn)行提取保留時(shí)間和色譜峰面積,每組試驗(yàn)重復(fù)測(cè)定的結(jié)果以回收率和相對(duì)標(biāo)準(zhǔn)偏差,用Microsoft Office Excel 2017軟件進(jìn)行數(shù)據(jù)處理。使用Origin 8.0繪制譜圖。
2 結(jié)果與分析
2.1 方法性能指標(biāo)
配制8種農(nóng)藥不同濃度的系列混合標(biāo)準(zhǔn)溶液,經(jīng)質(zhì)譜分析,8種農(nóng)藥的保留時(shí)間、定性離子、定量離子(見表1)色譜峰均獲得良好分離,峰形良好。標(biāo)準(zhǔn)色譜圖(有機(jī)氯類農(nóng)藥質(zhì)量濃度為1.0 mg/L的標(biāo)準(zhǔn)溶液,擬除蟲菊酯類農(nóng)藥質(zhì)量濃度為2.0 mg/L的標(biāo)準(zhǔn)溶液)如圖1所示。
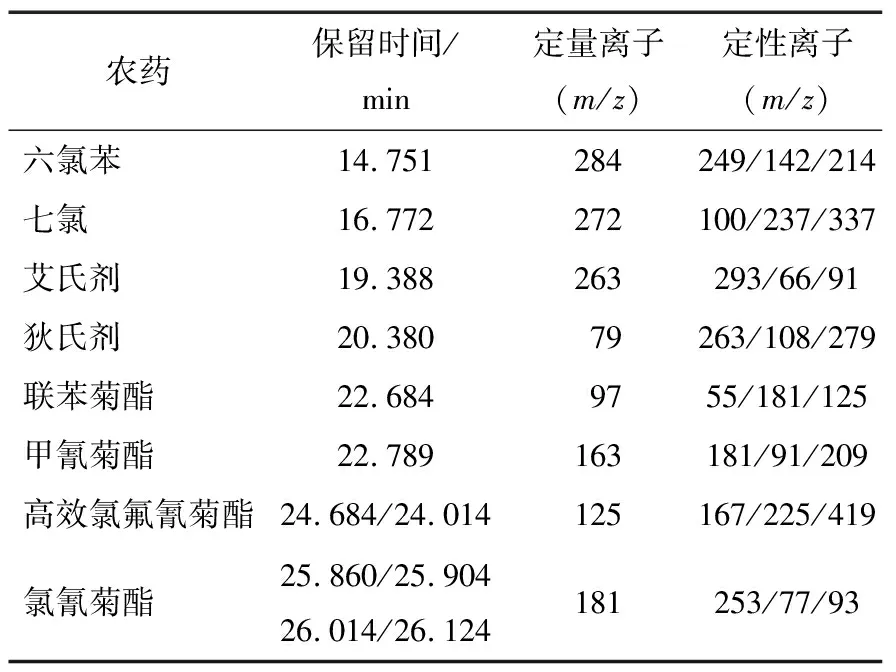
表1 8種農(nóng)藥保留時(shí)間和SIM掃描離子

1.六氯苯 2.七氯 3.艾氏劑 4.狄氏劑 5.聯(lián)苯菊酯 6.甲氰菊酯 7.高效氯氟氰菊酯 8.氯氰菊酯 有機(jī)氯類農(nóng)藥質(zhì)量濃度為1.0 mg/L 擬除蟲菊酯類農(nóng)藥質(zhì)量濃度為2.0 mg/L
2.2 濁點(diǎn)萃取優(yōu)化
2.2.1 表面活性劑種類以及用量的選擇 選擇Tween-20、Triton X-100和PEG 4000 3種表面活性劑作為萃取劑時(shí),8種農(nóng)藥的回收率。結(jié)果發(fā)現(xiàn),Tween-20和Triton-100為萃取劑時(shí),未能與反萃取溶劑異辛烷分層或分層不明顯,無法取上層清液進(jìn)樣分析;PEG 4000為萃取劑時(shí)可與反萃取劑經(jīng)離心有較好的分層,萃取效果好,故選用PEG 4000為萃取劑。考察不同用量的PEG 4000對(duì)回收率的影響,添加量分別設(shè)置為0.5,1.0,2.0,3.0 mL,回收率結(jié)果見圖2。當(dāng)PEG 4000(300 g/L)添加量為0.5~2.0 mL時(shí),8種農(nóng)藥的回收率均呈上升狀態(tài),添加量為2.0~3.0 mL時(shí),8種農(nóng)藥的回收率保持恒定,不再上升。因此,采用添加2 mL 300 g/L的PEG 4000進(jìn)行萃取。
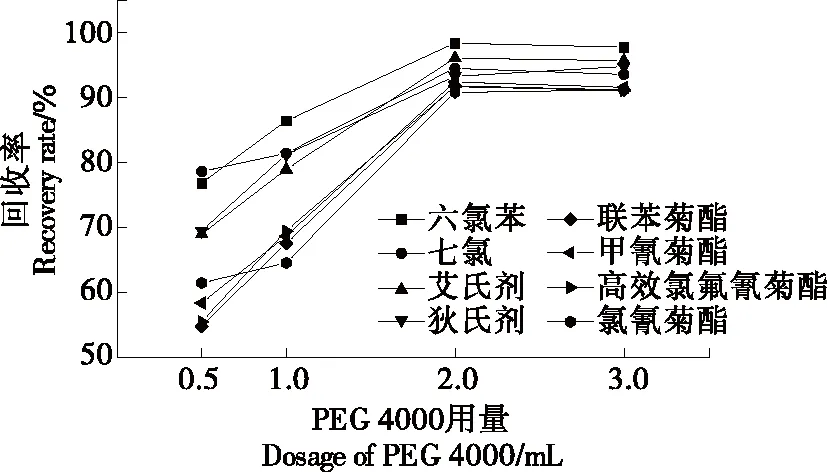
圖2 表面活性用量的選擇
2.2.2 鹽種類及用量的選擇 試驗(yàn)以無水Na2SO4為惰性鹽,考察了無水Na2SO4添加量(0.5~1.2 g)對(duì)濁點(diǎn)萃取的影響。由圖3可知:隨著無水Na2SO4添加量的增加,萃取回收率增大,當(dāng)無水硫酸鈉添加量達(dá)到1.0 g時(shí)回收率最好,此時(shí)溶液接近飽和,再增加無水硫酸鈉對(duì)回收率無影響。故試驗(yàn)中所用無水硫酸鈉用量為1.0 g。
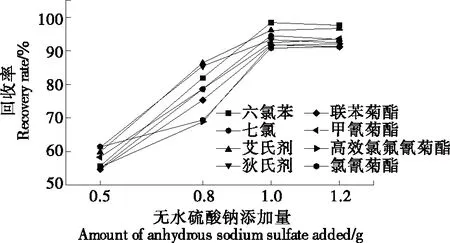
圖3 無水硫酸鈉添加量對(duì)8種農(nóng)藥回收率的影響
2.2.3 萃取溫度及水浴平衡時(shí)間的選擇 萃取過程中,較高的溫度和較長(zhǎng)的平衡時(shí)間并不會(huì)提高回收率,相反,較高的溫度和較長(zhǎng)的平衡時(shí)間會(huì)使待測(cè)物揮發(fā)、水解,從而造成損失[20]。樣品經(jīng)上述優(yōu)化處理后,分別在20,30,40,50 ℃水浴平衡20 min。由圖4可知,萃取溫度為20~40 ℃時(shí),8種農(nóng)藥的回收率均呈上升狀態(tài);萃取溫度高于40 ℃后,8種農(nóng)藥的回收率均呈下降趨勢(shì)。故試驗(yàn)采用的濁點(diǎn)萃取溫度為40 ℃。同時(shí)研究在40 ℃水浴下不同水浴平衡時(shí)間(10,15,20,30,40 min)對(duì)回收率的影響。由圖5可知,在40 ℃水浴中平衡20 min 時(shí)8種農(nóng)藥的回收率最好,較高的溫度和較長(zhǎng)的平衡時(shí)間并未提高回收率。
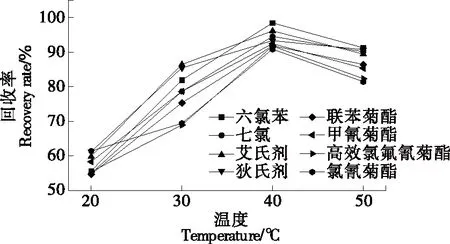
圖4 溫度對(duì)8種農(nóng)藥萃取回收率影響
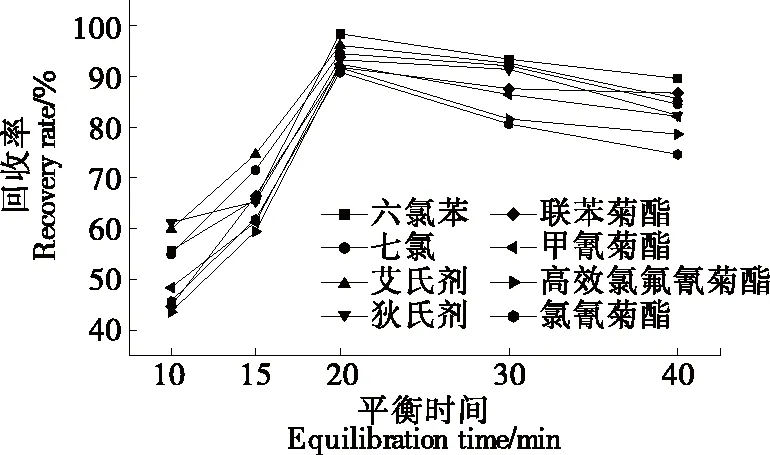
圖5 水浴平衡時(shí)間的選擇
2.3 反萃取條件優(yōu)化
表面活性劑相因其性質(zhì)黏稠直接進(jìn)入氣質(zhì)儀會(huì)造成污染,試驗(yàn)考慮選擇正己烷、石油醚、異辛烷作為反萃取溶劑,由于正己烷和石油醚本身的揮發(fā)性較強(qiáng),易揮發(fā)于超聲反萃取過程中,因此重現(xiàn)性較差,而異辛烷與表面活性劑不互溶,提取效果好,故選擇異辛烷作為反萃取劑。按上述前處理方法操作,研究異辛烷用量(100,150,200,300,500 μL)對(duì)回收率的影響,結(jié)果(見圖6)表明,采用200 μL異辛烷反萃取時(shí),回收率較高。
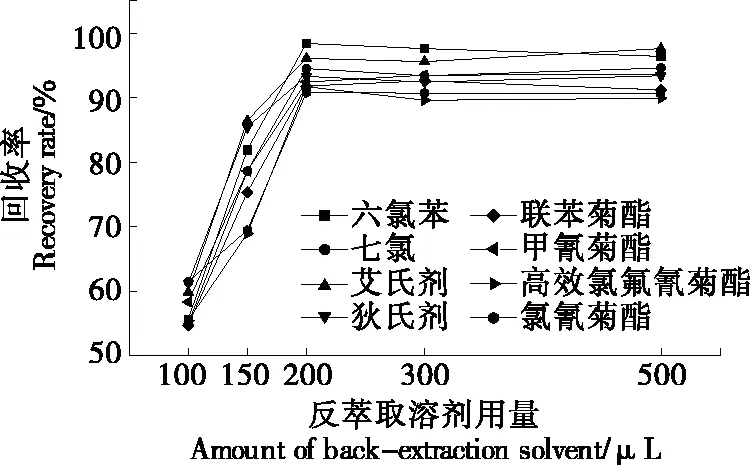
圖6 反萃取溶劑用量的選擇
2.4 方法學(xué)考察
2.4.1 線性范圍、相關(guān)系數(shù)和方法檢出限、定量限 用異辛烷將混合標(biāo)準(zhǔn)儲(chǔ)備液逐級(jí)稀釋成混合標(biāo)準(zhǔn)系列按上述優(yōu)化好的CPE-GC/MS法,檢測(cè)得到線性范圍、線性方程、相關(guān)系數(shù),見表2。

表2 CPE-GC/MS方法8種農(nóng)藥的工作曲線、線性相關(guān)系數(shù)、檢出限和定量限
試驗(yàn)采用在空白樣品中添加標(biāo)準(zhǔn)溶液按前處理后進(jìn)行檢測(cè),以最低濃度的加標(biāo)回收樣液計(jì)算信噪比(S/N),以S/N=3得出檢出限(LOD),S/N=10得出定量限(LOQ)。經(jīng)計(jì)算,確定該方法的檢出限為0.003~0.008 mg/L,定量限為0.01~0.02 mg/L。
2.4.2 加樣回收率和精密度試驗(yàn) 吸取10.0 mL空白茶飲料18份,按照試驗(yàn)優(yōu)化的前處理方法和色譜條件下,每個(gè)加標(biāo)濃度檢測(cè)6份,分別考察了低、中、高加標(biāo)濃度下8種農(nóng)藥的回收率,結(jié)果(見表3)表明,8種農(nóng)藥的平均回收率為78.3%~98.4%,相對(duì)標(biāo)準(zhǔn)偏差(RSD)2.7%~8.5%,該方法的精密度和準(zhǔn)確度均能達(dá)到滿意的結(jié)果,符合農(nóng)殘檢測(cè)方法確認(rèn)的要求。
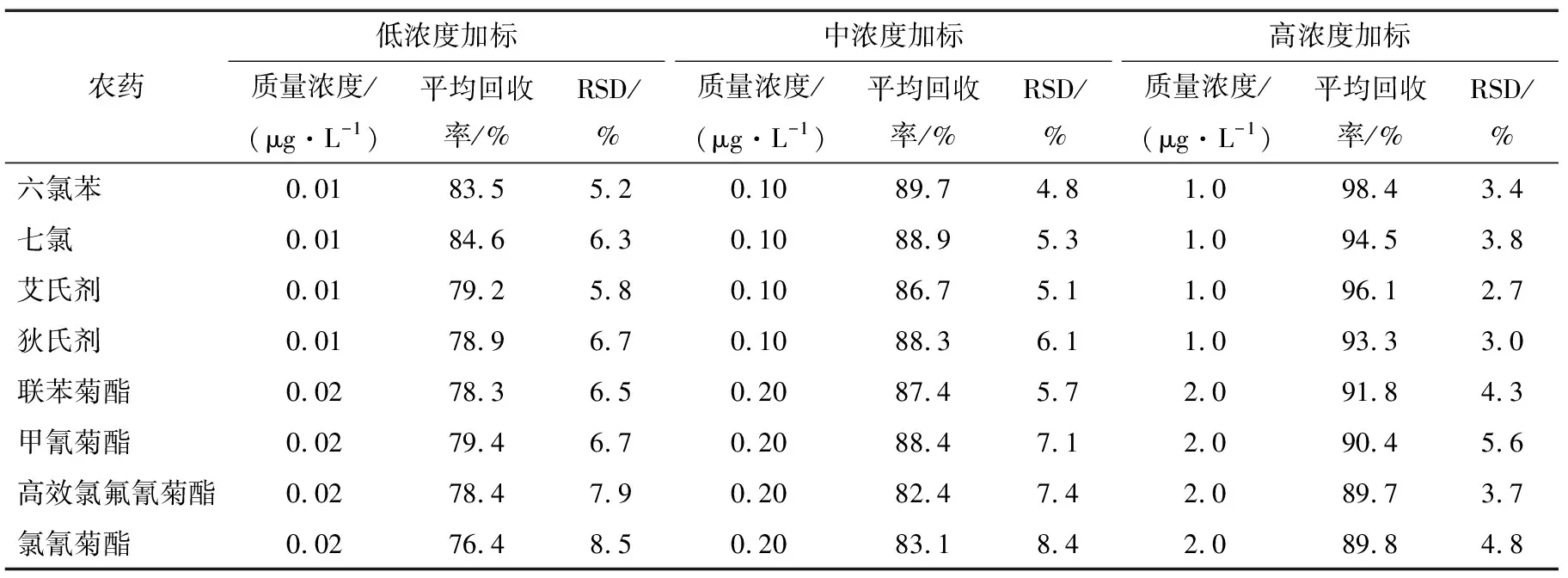
表3 3種不同加標(biāo)濃度下 8種農(nóng)藥的回收率和相對(duì)標(biāo)準(zhǔn)偏差
2.5 與已知方法的比較
將所建立的方法與文獻(xiàn)[3,5,9,11,20]報(bào)道的傳統(tǒng)方法從檢出限、定量限、精密度、回收率等方面進(jìn)行比較,分散固相萃取、分散液液微萃取、固相萃取、QuEChERS凈化等這些傳統(tǒng)的前處理方法會(huì)使用到大量的有機(jī)溶劑,會(huì)對(duì)試驗(yàn)人員造成損害,對(duì)環(huán)境造成污染。與其他方法相比,研究以非離子表面活性劑作為萃取劑快速有效地提取和預(yù)濃縮分離目標(biāo)物,再用微量的異辛烷進(jìn)行反萃取,該方法具有操作簡(jiǎn)單,高效,富集率高,且有機(jī)溶劑用量少,整體優(yōu)化了常規(guī)對(duì)茶飲料類液體飲料中農(nóng)殘的提取方法。傳統(tǒng)農(nóng)藥殘留檢測(cè)及預(yù)處理方法比較見表4。
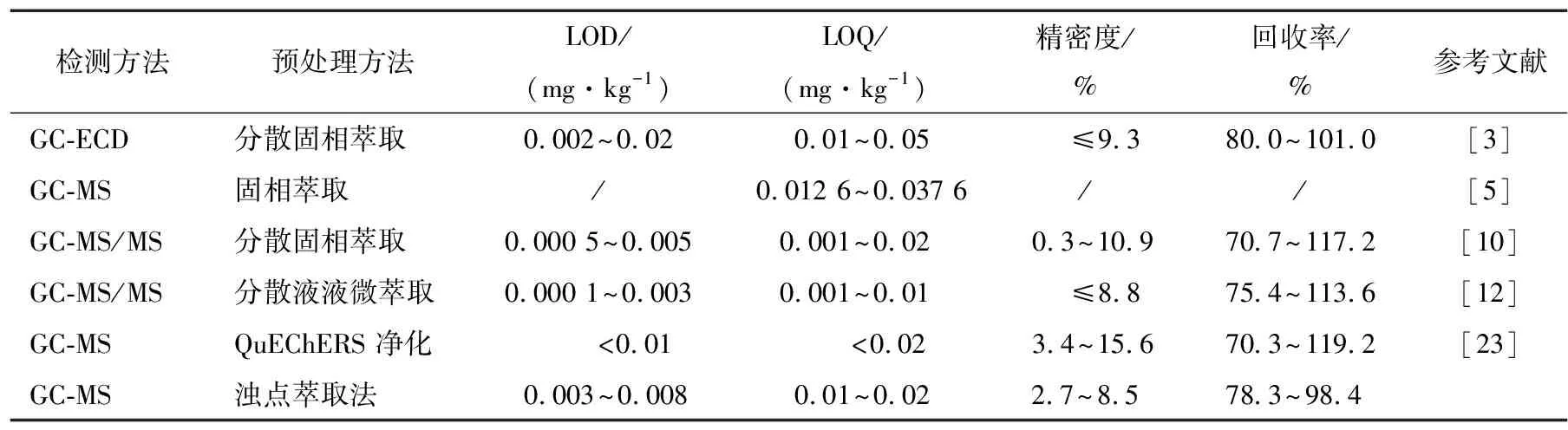
表4 與已知的農(nóng)藥殘留測(cè)定方法的比較
2.6 實(shí)際樣品的檢測(cè)
采集市場(chǎng)上不同品牌的6個(gè)茶飲料樣品按照建立的分析方法進(jìn)行農(nóng)藥殘留的測(cè)定,結(jié)果均為未檢出,符合食品安全要求。
3 結(jié)論
研究通過優(yōu)化表面活性劑種類及用量、鹽種類及用量、萃取溫度及平衡時(shí)間、反萃取溶劑種類及用量等前處理?xiàng)l件,建立了濁點(diǎn)萃取—異辛烷反萃取—?dú)庀嗌V質(zhì)譜法測(cè)定茶飲料中8種廣泛使用的擬除蟲菊酯和有機(jī)氯類農(nóng)藥(艾氏劑、六氯苯、氯氰菊酯、甲氰菊酯、溴氰菊酯等)農(nóng)殘的技術(shù)。該方法分離效果好,具有良好的線性關(guān)系,相關(guān)系數(shù)R均大于0.995,方法的檢出限為0.003~0.008 mg/kg,定量限為0.01~0.02 mg/kg,平均回收率78.3%~98.4%。與傳統(tǒng)分散固相萃取、QuEChERS 凈化等前處理方法相比,具有快速、簡(jiǎn)便、環(huán)保、高效、準(zhǔn)確,有機(jī)溶劑用量少等特點(diǎn),萃取過程中以表面活性劑水溶液代替有機(jī)試劑萃取目標(biāo)分離物,微量異辛烷作為反萃取溶劑,降低試驗(yàn)中大量有機(jī)溶劑的加入對(duì)環(huán)境的污染及試驗(yàn)人員身體的損害,適用于茶飲料類液體飲品多種農(nóng)藥殘留的測(cè)定。
猜你喜歡
中老年保健(2021年9期)2021-08-24 03:52:04
河北畫報(bào)(2021年2期)2021-05-25 02:07:46
中學(xué)生數(shù)理化(高中版.高考理化)(2020年2期)2020-04-21 05:33:04
兒童繪本(2020年5期)2020-04-07 17:46:30
兒童故事畫報(bào)(2019年5期)2019-05-26 14:26:14
Coco薇(2016年2期)2016-03-22 02:42:52
山東青年(2016年1期)2016-02-28 14:25:23
Coco薇(2015年1期)2015-08-13 02:47:34
小雪花·成長(zhǎng)指南(2015年7期)2015-08-11 15:03:12
小雪花·成長(zhǎng)指南(2015年4期)2015-05-19 14:47:56
