常染色體隱性遺傳性Alport綜合征誤診為IgA腎病1例報道并文獻復習
2023-05-04 09:18:16袁平鄭莎莎
中國生育健康雜志 2023年3期
袁平 鄭莎莎
病例資料
患兒,女,7歲,主因“間斷血尿、蛋白尿2年余”于2021年01月25日入本院。2018年11月06日患兒因“上呼吸道感染”4 d后出現尿色異常就醫,表現為全程深紅色尿,無尿頻、尿急、尿痛,無尿中沉渣及絮狀物,無水腫、少尿。當地醫院檢查:尿常規示紅細胞3101個/uL,尿蛋白2+;24 h尿蛋白定量0.337 g/d(19 mg/kg);補體C3、C4正常;ASO增高(583.96 IU/mL);腎功能(血肌酐31 μmol/L)、自身抗體譜、ANCA無異常。病程第5天當地完善腎穿活檢,病理結果免疫熒光:IgA 3+、IgG 2+、IgM 1+、C3 2+、C1q 1+、Fib -,IV型膠原α3、α5鏈染色腎小球基底膜和包曼氏囊均為陽性。光鏡:可見9個腎小球,未見腎小球球性硬化及節段性硬化,六胺銀(PASM)染色可見5個腎小球,未見腎小球球性硬化及節段性硬化;腎小球系膜細胞和基質輕度彌漫性增生,系膜區少許嗜復紅蛋白沉積,毛細血管襻開放,基底膜無明顯增厚,無釘突雙軌改變,無系膜插入,上皮下、內皮下無明顯嗜復紅蛋白沉積,未見白金耳樣結構,壁層上皮細胞無增生,未見新月體形成;腎小管上皮細胞顆粒變性,無明顯萎縮,腎間質無明顯炎癥細胞浸潤及纖維化,小動脈管壁無明顯病變。電鏡:腎小球基底膜彌漫性偏薄,厚度小于250 nm,足突部分融合,系膜區可見電子致密物沉積。病理診斷:輕度系膜增生性IgA腎病(M1E0S0T0)。結合腎臟病理,臨床診斷為IgA腎病(IgA nephropathy,IgAN),給予甲潑尼龍沖擊1療程,繼以醋酸潑尼松35 mg/d(2 mg/kg/d)口服并遞減,聯合賽可平250 mg Bid(30 mg/kg/d)、洛汀新5 mg Qd治療2年余,期間復查尿常規蛋白1+~2+,尿紅細胞30~滿視野/HP;尿蛋白定量0.24~0.31g/24 h。腎功能正常。現為進一步診治,特來本院。目前口服醋酸潑尼松10 mg/d,賽可平和洛汀新劑量同前。
既往、個人史無特殊。家族中有一哥哥,現年10歲,體健,未查過尿常規;父母體健,未查過尿常規。
入院查體:神清,精神反應好,輕度庫欣貌,咽無充血,扁桃體無腫大,心肺腹無異常,神經系統未見陽性體征。
入院后主要輔助檢查:全血生化示肌酐35.90 μmol/L,肝功能、電解質、心肌酶譜均正常;尿常規示蛋白質1+,紅細胞40~50/HP;尿蛋白/尿肌酐0.38 g/gcr;腎早期損傷指標示尿微量白蛋白60.90 mg/L,尿轉鐵蛋白和球蛋白正常;24 h尿蛋白定量 0.23 g;校正24 h肌酐清除率140 mL/min/1.73 m2;iPTH 14.45 pg/mL,25-OH-VitD 27.61 nmol/L;泌尿系超聲示雙腎實質回聲稍增強,肝膽胰脾未見異常,未探及腹腔積液。眼科會診示患兒雙眼視網膜顳側變薄,符合Alport綜合征(Alport syndrome,AS)眼部特征。耳鼻喉科行純音測聽提示雙耳大致正常聽力。患兒哥哥及父母尿常規蛋白均陰性,紅細胞3~5/HP。結合患兒臨床過程,按IgA腎病治療效果不佳,腎組織電鏡示腎小球基底膜彌漫性偏薄,家族中哥哥及父母均有鏡下血尿,考慮遺傳性腎小球腎炎不除外,二代測序完善家系遺傳性腎臟病相關基因變異分析,結果回報COL4A4基因存在2個位點雜合變異,c.5044C>G(p.R1682G,父源)和c.871-1G>A(母源),按照美國醫學遺傳學與基因組學學會(The American College of Medical Genetics and Genomics,ACMG)指南,變異位點分別初步判定為致病意義未明(PM2 +PM5+PP3)及疑似致病性變異(PVS+PM2),結合患兒臨床表現、家族史及藥物反應,明確診斷為“常染色體隱性遺傳性Alport綜合征”,見圖1、圖2。
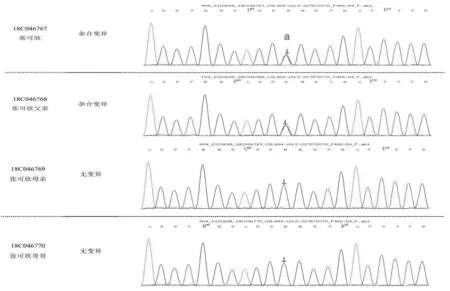
圖1 患兒家系的COL4A4基因測序圖1注:a為變異位點1,chr2:227872070,NM_0000 92;exon48,c.5044C>G(p.R1682G),為錯義變異
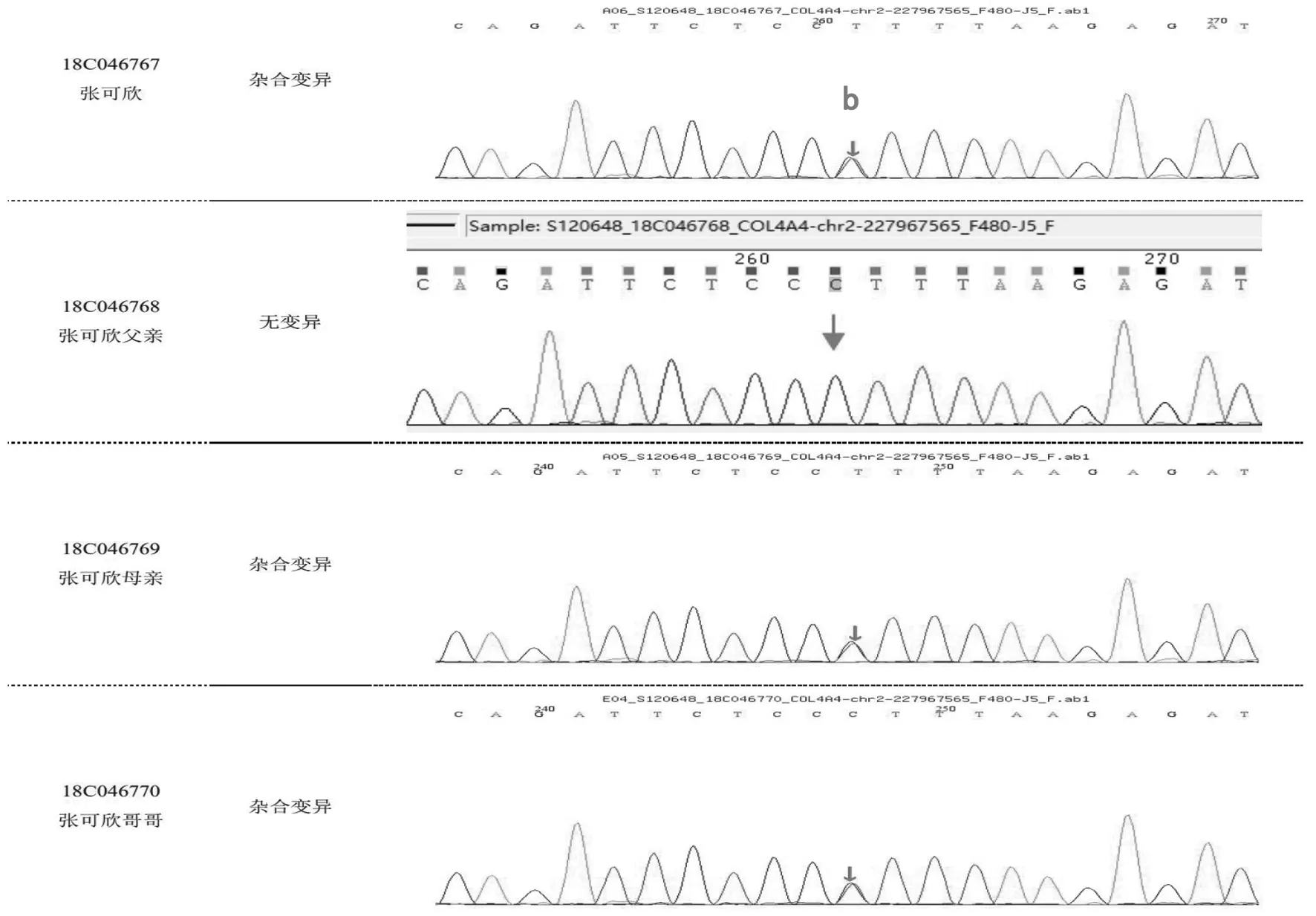
圖2 患兒家系的COL4A4基因測序圖變異位點2注:b為變異位點2,chr2:227872070,NM_0000 92;exon48,c.5044C>G(p.R1682G),為錯義變異。峰圖顯示的堿基有可能為被檢測堿基的反向互補序列,如:c.163G>A,峰圖可顯示為 G>A 或其反向互補序列 C>T
患兒診斷明確后,逐漸減停激素、賽可平,繼續洛汀新5 mg Qd口服,隨訪近1年,24 h尿蛋白定量(0.25~0.38 g)和腎功能(血肌酐32~36 μmol/L)較前無明顯變化,生長發育同正常同齡兒。
討論
IgAN是兒童最常見的原發性腎小球疾病之一,也是成人慢性腎臟疾病及終末期腎臟病的重要病因之一,主要在青年中好發,兒童也很常見。兒童年發病率為(0.03~4.5)/100 000[1]。IgAN的主要病理學特征是免疫熒光以IgA在腎小球沉積為主以及局部炎癥、系膜增生、腎小球纖維化等,其診斷基于主要依賴于腎組織免疫熒光染色結果。
AS是一種以血尿、蛋白尿和進行性腎功能衰竭為特征的慢性進展性腎臟疾病,可伴有高頻感音神經性耳聾和眼部病變,其診斷主要依賴基因變異分析或腎組織電鏡下腎小球基底膜特征性改變。IgAN和AS二者具有相同的臨床表現(血尿、蛋白尿、終末期腎病),特別是二者也可合并發生,臨床可能會發生誤診、漏診,甚至過度用藥誤治的情況。
IgAN臨床表現差異性大,起病形式包括無癥狀或輕微尿檢異常(包括鏡下血尿及少量蛋白尿)、持續蛋白尿及腎功能迅速惡化,一些患者沒有進展到終末期腎臟病(ESRD),甚至有報道稱一些患者自發緩解[2]。IgAN的經典表現是與黏膜感染相關的發作性肉眼血尿或持續性鏡下血尿。兒童IgAN有和成人類似的蛋白尿水平,但肉眼血尿更常見,血壓升高不明顯,腎功能保留較好。兒童比成人更常出現肉眼血尿,通常與上呼吸道感染有關。AS是因編碼Ⅳ型膠原蛋白α3、α4、α5鏈基因變異所致的遺傳性腎臟疾病,在臨床上表現為血尿、蛋白尿及進行性腎衰竭,部分患者合并耳聾和眼部改變[3],其遺傳方式有X連鎖顯性、常染色體隱性、常染色體顯性遺傳,其中以X連鎖顯性最常見,約占85%,主要為COL4A5的基因缺陷導致,常染色體隱性遺傳(ARAS)為COL4A3或 COL4A4基因缺陷所致,約占10%;常染色體顯性遺傳(ADAS),不到5 %,臨床非常罕見[4]。該病起病隱匿,早期病變表現輕微,僅為鏡下血尿,伴或不伴蛋白尿,腎功能異常多在學齡期甚至青春期后出現,聽力和視力損害也同樣出現較晚。
本例患兒以上呼吸道感染后發作性肉眼血尿起病,臨床符合IgAN特點,結合病初腎臟病理免疫熒光以IgA沉積為主,符合IgAN診斷,但規律激素聯合免疫抑制劑無明顯療效,尿蛋白定量無明顯變化,不符合兒童IgAN診斷常規病程。進一步檢查發現眼底AS特征性病變,且家族中哥哥、父母均鏡下血尿,考慮遺傳性腎小球腎炎不除外。基因變異證實COL4A4復合雜合變異(exon48:c.5044C>G p.R1682G,父源;exon15:c.871-1G>A,母源),診斷為常染色體隱性遺傳AS(COL4A4復合雜合變異)。
IgAN合并AS國內有病例報道,X連鎖顯性遺傳性AS多見[5-6]。已知正常健康人,腎組織活檢免疫熒光IgA沉積者約10%~30%[7-8],因此其他一些其他常見遺傳性腎小球腎炎通過腎組織活檢可能被誤診為IgAN。本例患兒,臨床經過激素聯合免疫抑制劑治療,尿蛋白水平無明顯下降趨勢,因此考慮其尿蛋白主要為原發病即常染色體隱性遺傳AS引起,與腎組織IgA沉積無明顯相關性,因此可以認為臨床被誤診、誤治或過度治療。提示在臨床工作中,以血尿起病的患兒很常見,既使通過腎活檢明確診斷為IgAN,但如果在后期的治療過程中出現病情多次反復或藥物治療效果欠佳,應積極詢問家族史、完善家系成員尿常規篩查,必要時相關基因變異分析以明確診斷,有無合并遺傳性腎小球腎炎或誤診可能。
AS的病理學常表現為系膜增生,時有IgA沉積,此外還有獨特的腎小球基底膜(GBM)改變,包括基底膜變薄和/或板層化。然而,在IgAN中也經常觀察到類似的GBM異常。這兩種疾病在與病毒感染相關時也會出現血尿、蛋白尿,有時還會出現大量血尿。因此,即使根據臨床和病理結果,也很難做出鑒別診斷。一項日本的回顧性研究[9]對5例同時伴有IgA沉積和GBM改變的患者進行了全面的基因篩查發現,其中4例被診斷為AS,1例為IgAN,伴有大量GBM改變,基因檢測結果為陰性。在4例被診斷為AS的患者中,腎活檢IgA在系膜區沉積在+至2+之間,未檢測到腎小球Gd-IgA1沉積,盡管系膜區IgA呈陽性;在1例IgA腎病,IgA沉積呈3+,并觀察到腎小球Gd-IgA1顯著沉積。提示反復血尿,治療效果不佳的病人,即使有典型的IgA腎病的臨床特點及常規病理結果,也難區分IgAN還是AS,還需進一步行基因檢測,以明確診斷AS。此外,Gd-IgA1染色有助于腎小球腎炎伴GBM異常和系膜IgA沉積的準確診斷,需要更多的分析來評估腎小球Gd-IgA1染色的效用。但國內絕大部分腎活檢免疫熒光均未行Gd-IgA1染色,值得注意的是當反復血尿和/或蛋白尿的病人行腎活檢免疫熒光提示IgA少量沉積時,需考慮是否為AS,并及時行基因檢測。在一項多中心對36例IgAN患兒進行Ⅳ型膠原蛋白的基因檢測的回顧性研究中發現[10],4例(4/36)患兒存在COL4A3變異,未檢出COL4A4及COL4A5變異,并認為COL4A3變異是導致IgAN嚴重發作的易感因素。已知IgA只是一種免疫熒光病理診斷,診斷時要結合臨床,已知部分正常人也可以有腎組織IgA沉積,因此,不是所有IgA沉積者均診斷為IgAN。全基因組關聯研究(GWAS)將某些血管緊張素(ACE)基因多態性確定為IgAN進展的獨立危險因素[11],在IgAN的發病機制中涉及多個基因,包括導致AS的變異基因[12-14]。
總之,本文首次在國內報導1例常染色體隱性遺傳性AS誤診、誤治為IgA腎病兒童例,提示臨床醫師凡是臨床過程特別是治療無效的原發性腎小球疾病如IgA腎病等,注意有無合并遺傳性腎小球疾病如AS可能,要意識到腎組織IgA沉積不代表一定就是IgA腎病,臨床過程一定要相符,特別是治療反應。凡是對于臨床治療反應欠佳的任何疾病,均應再次審視原發病診斷有無疑問。
