不同病理類型甲狀腺癌共同調控網絡的轉錄組學整合研究△
2023-05-22 03:38:30謝麗君
癌癥進展 2023年6期
關鍵詞:分析
謝麗君
昆明醫科大學第一附屬醫院核醫學科,昆明 653002
甲狀腺癌是全球常見的惡性腫瘤,甲狀腺乳頭狀癌(papillary thyroid carcinoma,PTC)是主要的甲狀腺癌類型,治愈率超過90%;相反,濾泡狀甲狀腺癌(follicular thyroid carcinoma,FTC)易發生血行轉移且復發率高,甲狀腺未分化癌(anaplastic thyroid carcinoma,ATC)、甲狀腺髓樣癌(medullary thyroid carcinoma,MTC)是最具侵襲性的內分泌惡性腫瘤,ATC 和MTC 對放射性碘消融和傳統化療無效。因此,識別PTC、ATC、MTC 和FTC 間主調節因子的差異和共同調控的信號網絡可能對甲狀腺癌的治療有重要意義。
近年來,多項研究闡明了不同病理類型甲狀腺癌的遺傳復雜性。Yoo 等[1]比較了12 例晚期分化型甲狀腺癌(differentiated thyroid carcinoma,DTC)和13 例ATC 患者的mRNA 表達情況發現,異常的甘油脂類代謝、脂肪酸代謝、CDKN2A基因表達與甲狀腺癌的侵襲性明顯相關。Nicolson 等[2]對17例FTC 和4 例正常甲狀腺組織的轉錄組進行測序發現,組蛋白去乙酰化酶1(histone deacetylase 1,HDAC1)是一種能在腫瘤侵襲前發揮增強表達作用的組蛋白修飾物。盡管既往已取得了重要進展,但PTC、ATC、MTC 和FTC 間的共同調控網絡和特異性調控因子仍然未知。
本研究從GSE27155、GSE29265 和GSE53157共3 個數據集的組織樣本中篩選基因表達譜,共包括9 例ATC、43 例PTC、2 例MTC 和4 例FTC 患者的數據,通過PTC 分別與FTC、ATC 和MTC 比較的交集來確定差異表達基因(differential expressed gene,DEG);此外,采用基因富集分析注釋4 種不同病理類型甲狀腺癌共同特異基因的生物學功能和信號通路;構建轉錄因子調控網絡以確定甲狀腺癌的共同主調控因子,并分析其與甲狀腺癌患者預后的關系,現報道如下。
1 資料與方法
1.1 DEG 篩選
通過基因表達綜合數據庫(Gene Expression Omnibus,GEO)進行DEG 篩選,在GEO 數據庫中以“thyroid cancer”“Homo sapiens”“tissue”為關鍵詞搜索基因序列,排除微小RNA、線粒體DNA、非體內甲狀腺癌組織取材標本等相關數據,最終選取GSE27155、GSE29265 和GSE53157 共3 個數據集,其中GSE27155 的數據來自26 例PTC 和2 例MTC 患者,GSE29265 的數據來自10 例PTC 和9 例ATC 患者,GSE53157 的數據來自7 例PTC 和4 例FTC 患者。利用基因芯片差異分析軟件GEO2R(https://www.ncbi.nlm.nih.gov/geo/geo2r/)分析各個數據集,選取P<0.05、|logFC|>1 的基因為候選差異基因,最終將3 個數據集中選取的差異基因取交集,篩選出DEG。利用微生信在線分析軟件(http://www.bioinformatics.com.cn/)對篩選出的DEG 進一步分析以篩選出共同表達的差異基因。
1.2 基因本體(gene ontology,GO)功能注釋和京都基因與基因組百科全書(Kyoto Encyclopedia of Genes and Genomes,KEGG)通路富集分析
利用2021 更新版生物學信息注釋及可視化數據庫(the database for annotation,visualization and integrated discovery,DAVID)(https://david.ncifcrf.gov/tools.jsp)中在線分析工具以人源基因為背景進行GO 功能注釋和KEGG 通路富集分析,設定P<0.05。
1.3 蛋白相互作用網絡分析
利用String 數據庫(https://cn.string-db.org/)構建蛋白質相互作用網絡,并采用Cytoscape 3.7.2 進行可視化分析,通過CytoHubba 插件對構建的蛋白質相互作用網絡進行關聯度分析。通過分析網絡結構,根據關聯積分值,獲得整個網絡中可能形成的蛋白簇和關鍵節點蛋白,并在Cytoscape 中進行可視化。關鍵節點篩選標準為最大團中心性(maximal clique centrality,MCC)算法,同時采用Mcode 插件進行蛋白簇和關鍵節點蛋白分析,隨后采用ClueGO 插件(http://apps.cytoscape.org/apps/cluego)對CytoHubba 生成的模塊進行通路相互作用網絡構建。
1.4 轉錄因子的預測
通過Cytoscape 3.7.2 軟件中的iRegulon 插件對1.3 中獲得的關鍵節點蛋白進行轉錄因子的預測。預測轉錄因子時,參數被設置為默認值,此外,選擇標準化富集分數(normalized enrichment score,NES)>5.0 的轉錄因子作為重要因子的調控網絡。
1.5 不同病理類型甲狀腺癌共同轉錄因子的Kaplan-Meier 分析
為評估1.4 中獲得的轉錄因子對甲狀腺癌患者預后的預測價值,使用Kaplan-Meier plotte 計算表達值的最佳臨界值[3],將來自癌癥基因組圖譜(The Cancer Genome Atlas,TCGA)(https://portal.gdc.cancer.gov/)的353 例甲狀腺癌組織分為高表達組與低表達組,進行Kaplan-Meier 生存分析(www.kmplot.com),組間比較采用Log-rank 檢驗。對于Kaplan-Meier 曲線,P值和95%CI 的風險比(hazard ratio,HR)通過Log-rank 檢驗和多因素Cox 回歸分析得出,以P<0.05 為差異有統計學意義,其中不同組間HR 代表高表達組相對于低表達組的風險系數,若HR>1 代表該基因為危險因素,若HR<1 則代表該基因為保護因素。
2 結果
2.1 不同病理類型甲狀腺癌DEG 的篩選
通過對GSE27155、GSE29265 和GSE53157 共3個數據集中PTC 與MTC、PTC 與ATC、PTC 與FTC比較分析,分別確定了2394、1104 和2805 個獨立的DEG,共同交集篩選獲得191 個共同表達的DEG,其中上調基因143 個,下調基因48 個。(圖1)
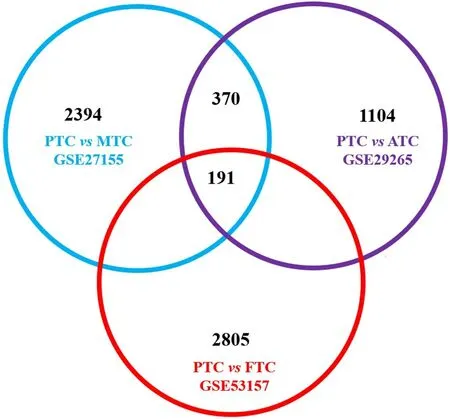
圖1 不同病理類型甲狀腺癌的DEG篩選圖
2.2 GO 功能注釋分析
選取2.1 中所篩選出的共同表達的DEG,利用DAVID 在線分析工具共得出96 項重要富集的基因功能,其中生物進程主要富集于細胞形態調控、細胞黏附的負調節、細胞連接組件的正向調節等;細胞成分主要富集于細胞前緣、絲狀膜、外側質膜等;分子功能主要富集于其他取代磷酸基團的磷酸轉移酶活性、Poly(A)結合、細胞間黏附介質活性等。(圖2)
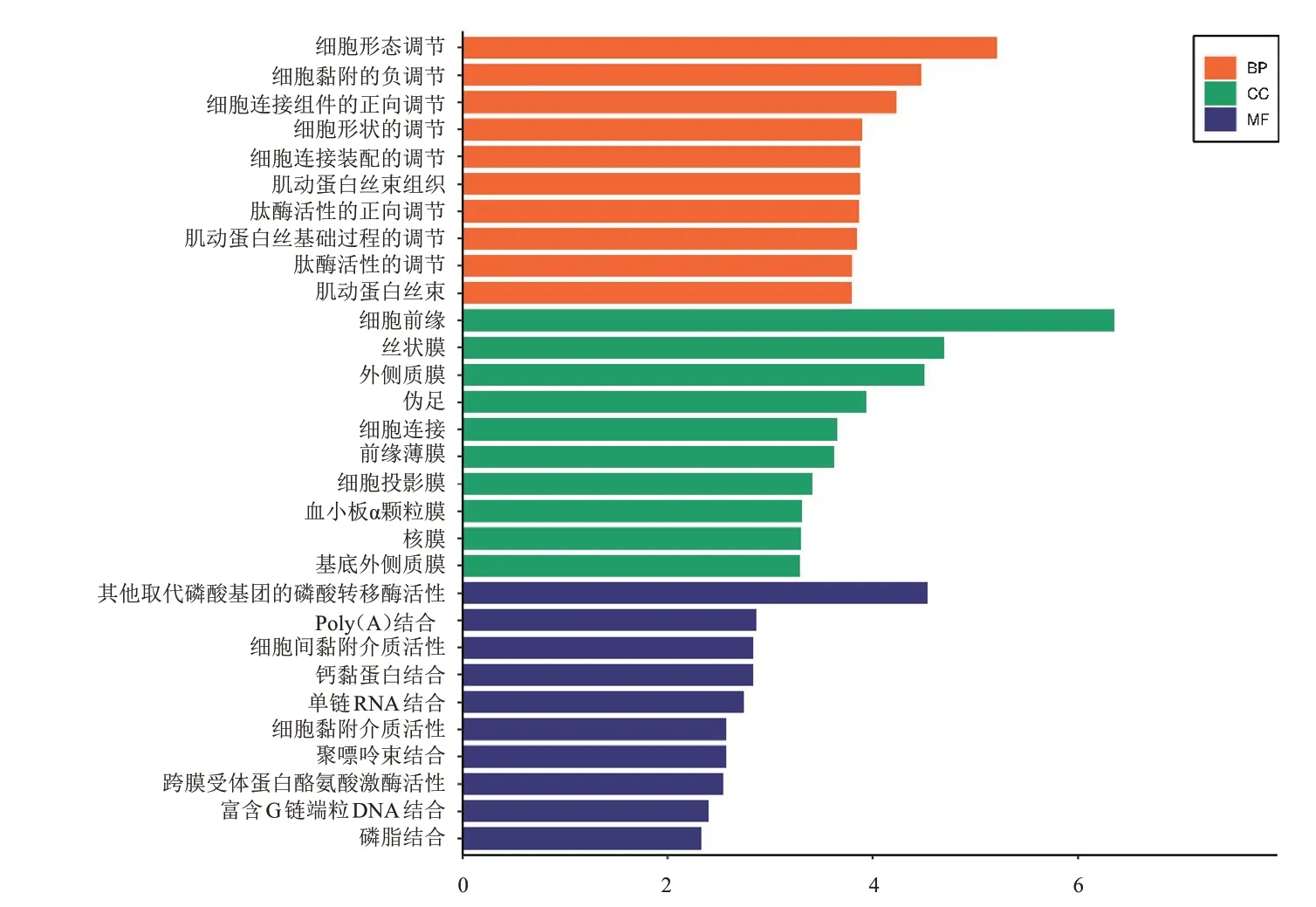
圖2 191個共同表達的DEG的GO功能注釋分析
2.3 KEGG 通路富集分析
選取2.1 中所篩選出的共同表達的DEG,利用DAVID 在線分析工具共得出重要富集的5 條KEGG 通路,主要包括堿基切除修復、膦酸鹽和次膦酸鹽代謝、致病性大腸桿菌感染、腫瘤中的蛋白聚糖、細胞黏附分子通路。涉及的主要基因包括多聚二磷酸腺苷核糖聚合酶4[poly(ADP-ribose)polymerase 4,PARP4]、8-氧鳥嘌呤DNA 糖苷酶(8-oxoguanine DNA glycosylase,OGG1)、DNA 聚合酶δ2 輔助亞基(DNA polymerase delta 2,accessory subunit,POLD2)、膽堿磷酸轉移酶1(choline phosphotransferase 1,CHPT1)、膽堿/乙醇胺磷酸轉移酶1 (choline/ethanolamine phosphotransferase 1,CEPT1)、連接蛋白(claudin,CLDN)3、肌球蛋白Ⅹ(myosin Ⅹ,MYO10)、FAS、肌球蛋白重鏈10(myosin heavy chain 10,MYH10)、CLDN1、促分裂原活化的蛋白激酶12(mitogen-activated protein kinase 12,MAPK12)、erb-b2 受體酪氨酸激酶3(erb-b2 receptor tyrosine kinase 3,ERBB3)、多配體蛋白聚糖4(syndecan 4,SDC4)、整合素β3(integrin subunit beta 3,ITGB3)、MET、鈣黏蛋白3(cadherin 3,CDH3)和蛋白酪氨酸磷酸酶受體M(protein tyrosine phosphatase receptor type M,PTPRM)。(圖3)
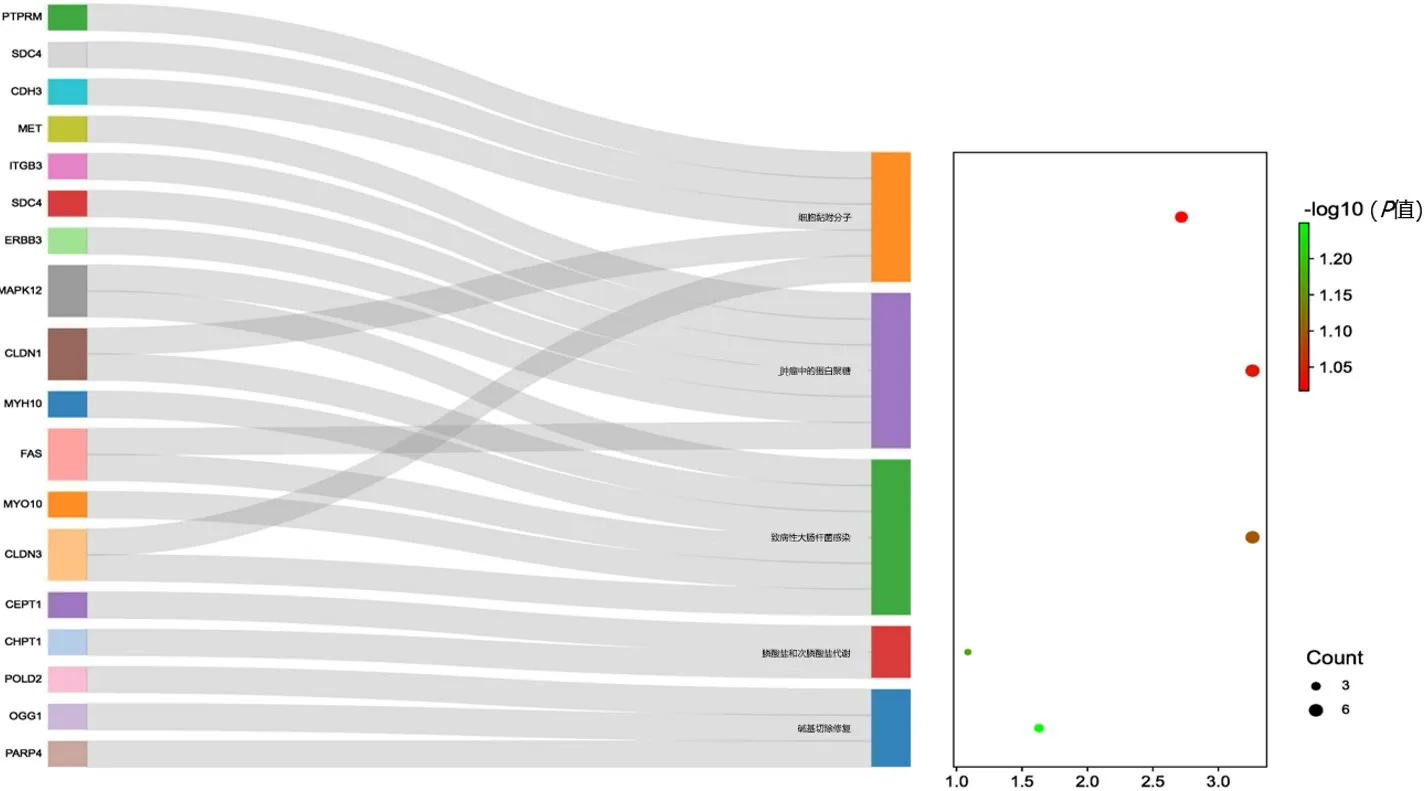
圖3 191個共同表達的DEG的KEGG通路富集分析
2.4 蛋白相互作用網絡可視化分析
基于前述的DEG 富集通路的蛋白構建相互作用網絡,對蛋白-蛋白相互作用進行可視化分析,結果顯示,甲狀腺癌共同表達的DEG 形成的蛋白相互作用網絡中含有24個下調基因及97個上調基因,并共同富集于重要的蛋白-蛋白相互作用通路上。
CytoHubba 插件的MCC 分析顯示有24 個節點、26 條邊,主要富集在蛋白質二硫鍵異構酶活性、鳥苷酸交換因子活性的負調節、磷脂酸磷酸酶活性的調節、富含脯氨酸區域結合、核纖層蛋白結合、磷脂酰肌醇3-激酶催化亞單位結合、SUMO 接合酶活性、細胞骨架-核膜錨定活性,關鍵基因包括凝溶膠蛋白(gelsolin,GSN)、RAS p21 蛋白激活因子(RAS p21 protein activator 1,RASA1)、真核生物翻譯起始因子2α亞單位(eukaryotic translation initiation factor 2 subunit alpha,EIF2S1)、UBX 結構域蛋白7(UBX domain protein 7,UBXN7)、ITGB3、Yes 相關轉錄調控因子1(Yes1 associated transcriptional regulator 1,YAP1)、脂蛋白1(lipin 1,LPIN1)、核膜含血影蛋白重復蛋白2(spectrin repeat containing nuclear envelope protein 2,SYNE2)、泛素結合酶E2C 結合蛋白E2I(ubiquitin conjugating enzyme E2I,UBE2I)、肌營養不良蛋白(dystrophin,DMD)。(圖4A)
Mcode 模塊1 分析包括6 個節點、6 條邊,主要富集在單酚單加氧酶活性的調節和非別肽酶活性的正調節信號通路,關鍵基因包括腫瘤關聯鈣信號轉導因子2(tumor associated calcium signal transducer 2,TACSTD2)、CLDN3、CLDN1、CDH3。(圖4B)
Mcode 模塊2 分析包括27 個節點、53 條邊,主要富集于二酰甘油膽堿轉移酶活性、磷脂酰肌醇轉移酶活性、富含G 鏈的端粒DNA 結合、錯配修復、細胞骨架-核膜錨定活性、磷脂酰磷酸酶活性調節通路,關鍵基因包括磷脂酸胞苷酸轉移酶1(CDP-diacylglycerol synthase 1,CDS1)、CEPT1、LPIN1、CHPT1、SYNE2、端粒重復序列結合因子2(telomeric repeat binding factor 2,TERF2)、端粒重復序列結合因子2 結合蛋白(TERF2 interacting protein,TERF2IP)、POLD2。(圖4C)
Mcode 模塊3 分析包括9 個節點、10 條邊,主要富集于磷脂酰肌醇-5-磷酸結合、磷脂酰肌醇磷酸激酶活性通路,關鍵基因包括磷脂酰肌醇-4-磷酸3-激酶催化亞單位2α(phosphatidylinositol-4-phosphate 3- kinase catalytic subunit type 2 alpha,PIK3C2A)、分揀微管連接蛋白5(sorting nexin 5,SNX5)、腫瘤蛋白D52 樣蛋白1(tumor protein D52 like 1,TPD52L1)。(圖4D)
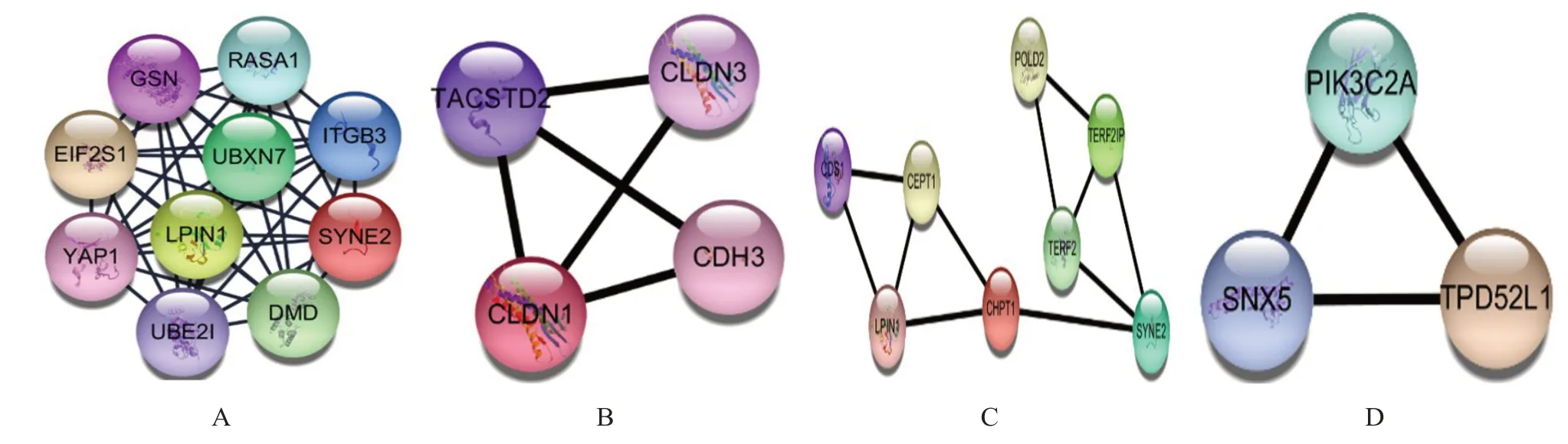
圖4 蛋白相互作用網絡的模塊分析和基因功能注釋圖
2.5 主要轉錄因子和網絡預測
使用iRegulon 插件對結果2.4 中的25 個關鍵基因進行轉錄因子預測,結果顯示,有14 個重要的轉錄因子高度富集于蛋白網絡中,包括EBF 轉錄因子1(EBF transcription factor 1,EBF1)、髓樣鋅指蛋白1(myeloid zinc finger 1,MZF1)、E2F 轉錄因子1(E2F transcription factor 1,E2F1)、信號轉導與轉錄激活因子(signal transduction and activator of transcription,STAT)6、核因子κB 亞基(nuclear factor kappa B subunit,NFKB)1、NFKB2、RELA、churchill結構域蛋白1(churchill domain containing 1,CHURC1)、銜接因子相關蛋白復合體3β1(adaptor related protein complex 3 subunit beta 1,AP3B1)、含鋅指和BTB 結構域7A(zinc finger and BTB domain containing 7A,ZBTB7A)、Ikaros 家族鋅指蛋白2(IKAROS family zinc finger 2,IKZF2)、上游轉錄因子2 與c-fos 相互作用(upstream transcription factor 2,c-fos interacting,USF2)、STAT1、ovo 樣鋅指蛋白2(ovo like zinc finger 2,OVOL2),NES=5.119。對應重要的靶基因包括細胞分裂周期14A(cell division cycle 14A,CDC14A)、Rho GTP 酶活化蛋白44(Rho GTPase activating protein 44,ARHGAP44)、倒置形成素蛋白2(inverted formin 2,INF2)、磷酸酶和肌動蛋白調控因子2(phosphatase and actin regulator 2,PHACTR2)、碳水化合物磺基轉 移 酶 15(carbohydrate sulfotransferase 15,CHST15)、CD47、UBE2I。
2.6 關鍵轉錄因子與甲狀腺癌患者預后的關系
評估2.5 中共同轉錄因子與甲狀腺癌患者預后的關系,以掌握甲狀腺癌的重要調節因子,使用Kaplan-Meier plotter 法計算上述基因高表達組與低表達組甲狀腺癌患者的無復發生存率(relapse-free survival,RFS),結果顯示,E2F1(HR=6.51,P=0.003)、NKFB1(HR=4.13,P=0.000)和NFKB2(HR=2.35,P=0.029)均是甲狀腺癌患者預后的危險因素,IKZF2(HR=0.27,P=0.023)和MZF1(HR=0.31,P=0.002)均是甲狀腺癌患者預后的保護因素。(圖5)
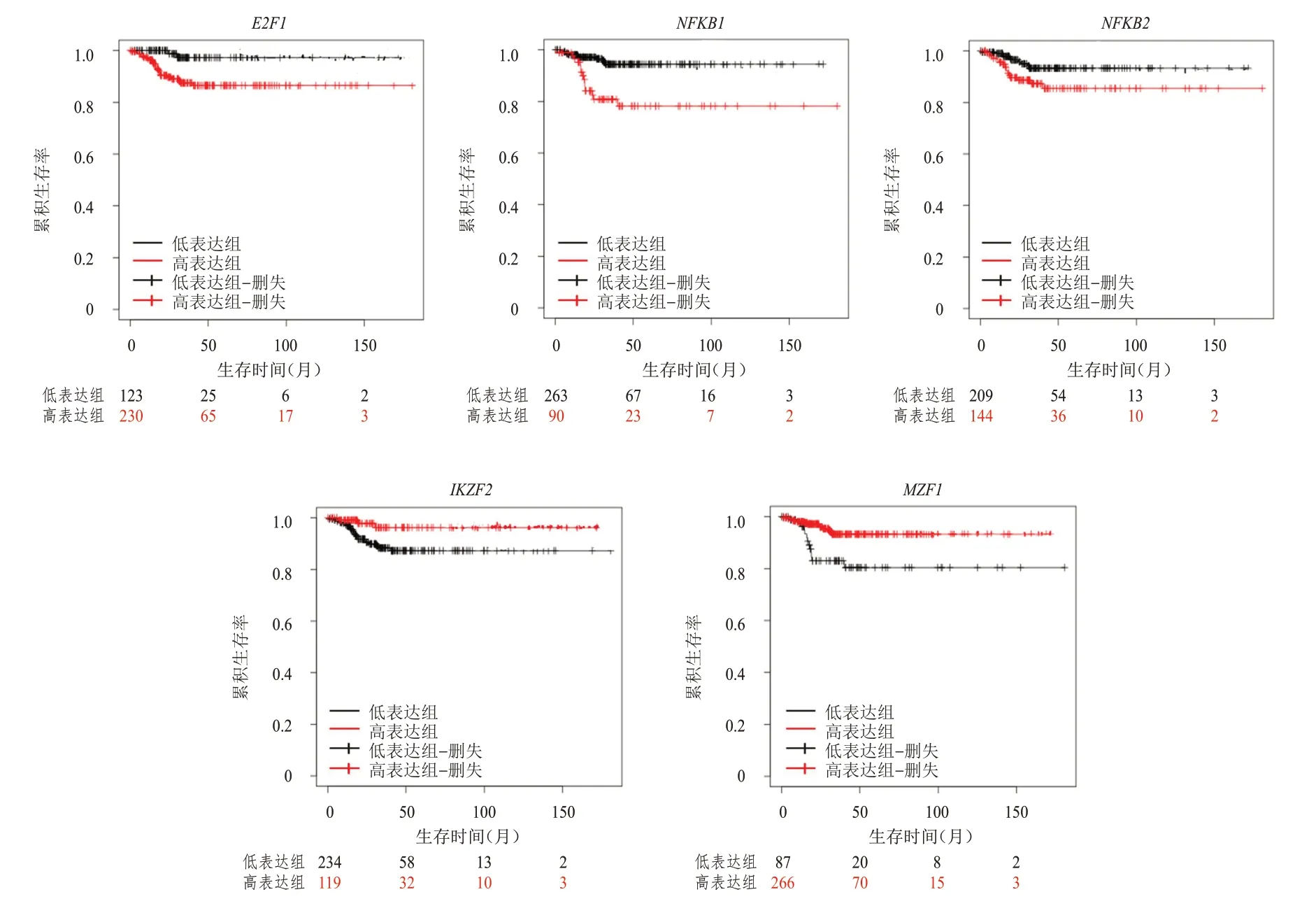
圖5 不同轉錄因子表達情況甲狀腺癌患者的RFS曲線
3 討論
不管是預后良好的PTC,還是預后極差的ATC和MTC,以及易發生血行轉移或遠處轉移的FTC,診療的相關問題已引起臨床醫師和研究者的高度關注。既往學者一直利用不同的技術關注不同病理類型甲狀腺癌的基因組或轉錄組改變[4]。但由于ATC 和MTC 的罕見性和異質性常導致不同研究結果出現誤差。本文從3 個獨立數據集中生成了一個大數據集以探索PTC、FTC、ATC 及MTC 間調控網絡的差異和共同存在的重要轉錄因子。
對FTC、ATC 及MTC 特定DEG 的識別可全面了解腫瘤的侵襲性。本研究通過比較發現,與PTC組織相比,FTC、ATC、MTC 組織中均存在獨特表達的基因。基因集富集分析發現,細胞周期、細胞有絲分裂、細胞增殖、免疫信號失調與不同病理類型甲狀腺癌發生發展密切相關。重要模塊分析和基因功能注釋分析也表明關鍵模塊也與細胞周期、細胞增殖有關。
有研究指出,細胞周期基因在甲狀腺癌中被高度激活[5-6],本研究提供了重要模塊轉錄因子調控網絡的預測,確定EBF1、MZF1、E2F1、STAT6、NFKB1、NFKB2、RELA、CHURC1、AP3B1、ZBTB7A、IKZF2、USF2、STAT1、OVOL2基因均是細胞周期和免疫調節相關模塊的潛在主調控因子。研究顯示,E2F1對控制G1/S 期基因表達至關重要[7-8]。本研究分析結果顯示,E2F1是甲狀腺癌中重要的特異因子,與Yang 等[9]的研究結果相似。此外,本研究證實了E2F1的潛在轉錄靶點可能是腫瘤細胞周期失調的機制。研究表明,轉錄因子NFKB可協同E2F靶基因的轉錄并調節細胞周期[10],NFKB的異常表達與多種腫瘤有關,其中就包括甲狀腺癌[11-12]。IKZF2是造血特異性轉錄因子,參與調節淋巴細胞發育,可控制細胞的自我更新和分化等功能[13],也具有腫瘤抑制因子的特性[14]。本研究同樣發現,IKZF2是甲狀腺癌的重要轉錄因子之一,但IKZF2如何在不同的細胞類型中發揮不同的作用尚不清楚。多數腫瘤患者死亡是由于腫瘤細胞轉移到其他器官,而侵襲是轉移形成的先決條件,有研究指出,MZF1是致癌因子,是驅動病變惡性轉化的重要節點[15]。本研究結果顯示,MZF1是甲狀腺癌患者預后的保護因素,表明其能調控腫瘤中特定靶基因的激活,但對于MZF1與甲狀腺癌全基因組相關性的研究仍待進一步深入研究。本研究篩選出的GSN、RASA1和EIF2S1等25 個關鍵基因的表達具有潛在的促進甲狀腺癌發生發展的作用。由此可見,E2F1、NKFB1、NFKB2、IKZF2、MZF1主調控因子及其潛在靶點所形成復雜的細胞周期基因調控網絡可能是治療效果差或易復發甲狀腺癌快速進展的原因。
綜上所述,本研究整合轉錄組學研究揭示了不同病理類型甲狀腺癌共同的惡性特征及相關性,E2F1、NKFB1、NFKB2、IKZF2、MZF1被認為是調控網絡中共同表達的DEG 的主要調控因子,這為甲狀腺癌的惡性機制提供新的見解。但本研究仍存在不足之處,未來仍需考慮一些因素如本文中的3個數據集中的樣本不對稱性及惡性程度高的甲狀腺癌樣品量少所帶來的潛在影響。盡管存在局限性,但這些發現仍可支持與甲狀腺癌相關的主要調控因子和網絡在未來研究中的特殊價值。
猜你喜歡
現代畜牧科技(2021年9期)2021-10-13 06:39:14
民用飛機設計與研究(2020年4期)2021-01-21 09:15:02
電子制作(2018年18期)2018-11-14 01:48:24
山東工業技術(2016年15期)2016-12-01 05:31:22
當代經濟研究(2016年5期)2016-12-01 03:12:05
現代農業(2016年5期)2016-02-28 18:42:46
出版與印刷(2016年3期)2016-02-02 01:20:11
中國中醫藥現代遠程教育(2014年11期)2014-08-08 13:23:44
華北水利水電大學學報(社會科學版)(2014年3期)2014-04-16 04:38:31
終身教育研究(2014年5期)2014-02-28 01:23:06
