基于活性位點調控的黃鐵礦浮選抑制研究進展
2023-07-06 01:01:16張晚佳陶黎明曹建孫偉高志勇
中南大學學報(自然科學版) 2023年5期
張晚佳,陶黎明,曹建,孫偉,高志勇
(1. 中南大學 資源加工與生物工程學院,湖南 長沙,410083;2. 中南大學 戰略含鈣礦物資源清潔高效利用湖南省重點實驗室,湖南 長沙,410083;3. 中南大學 湖南省關鍵金屬礦產資源高效清潔利用國際聯合研究中心,湖南 長沙,410083)
黃鐵礦(FeS2)是地殼中分布最廣的硫化物,也是提取硫和制造硫酸的主要礦物原料[1-2]。黃鐵礦常常作為脈石礦物與黃銅礦(CuFeS2)、方鉛礦(PbS)、閃鋅礦(ZnS)及金銀貴金屬礦物緊密共生,成為上述礦物高效利用的最大障礙[2-4]。如果不能及時分離黃鐵礦與其他有價礦物,不僅會降低精礦產品的回收率和品位,影響企業的經濟效益,而且給后續冶煉等環節造成不利影響[1]。黃鐵礦在冶煉過程中會釋放大量有害氣體如SO2和SO3,進而引發酸雨,給周邊水體和大氣環境帶來嚴重危害[5]。因此,不論是從經濟角度還是從環境保護角度出發,通過礦物浮選實現黃鐵礦與其他礦物的高效分離具有重要意義。
研究表明,黃鐵礦表面斷裂鍵密度從大到小依次為{111}、{311}、{110}、{021}、{001}晶面,而層間間距從大到小依次為{111}、{001}、{021}、{110}、{311}晶面[6]。由于礦物主要沿著具有較大層間距和少量斷裂鍵的方向解理,因此,具有最小的斷裂鍵密度和第二大的層間間距的{001}晶面是黃鐵礦最常見的解理面[6-7]。黃鐵礦晶體中包含以Fe—S 為主的離子鍵和以S—S 為主的共價鍵,前者的鍵能大于300 kJ/mol,而后者的鍵能較低,為 245 kJ/mol[8]。當黃鐵礦被破碎和研磨時,Fe—S鍵和S—S 鍵被破壞,暴露出Fe2+,這是黃鐵礦表面活性的根源,也是浮選藥劑在吸附時的主要目標位點。
在浮選中,調控上述活性位點是改變黃鐵礦表面反應活性的主要途徑。在加入捕收劑之前,提前加入能占據黃鐵礦表面活性位點并提高表面親水性的抑制劑,是降低黃鐵礦回收率的主要途徑之一。抑制劑的親固基團通過吸附作用牢牢抓住黃鐵礦表面的活性位點,同時親水基團與水分子緊密結合,協同阻止黃鐵礦上浮,最終實現礦物分選。上述過程通過抑制劑的預吸附來掩蓋黃鐵礦表面的活性位點,以降低其與捕收劑的鍵合概率,屬于間接抑制方法。而通過氧化作用來轉化黃鐵礦表面的活性位點則是抑制黃鐵礦的直接途徑。現階段氧化抑制主要包括磨礦氧化抑制、微生物氧化抑制和無機物氧化抑制等,其優勢在于無需顧慮后續其他浮選藥劑的競爭吸附。另外,黃鐵礦自身的導電性為轉化活性位點提供了便利。通過氧化還原反應將黃鐵礦表面的Fe2+活性位點氧化為鐵的氫氧化物和氧化物,同時親水氧化產物形成保護層,阻礙黃鐵礦和捕收劑及氣泡的結合上浮[9]。黃鐵礦表面具有電子空穴,易于傳導電子[2,10],使其具有半導體性質及參與光電反應的巨大潛能[10]。因此,借助光電化學技術定向降低黃鐵礦表面反應活性是提高黃鐵礦抑制效率的潛在研究方向之一。本文基于掩蓋或轉化活性位點的角度綜述了近年來黃鐵礦抑制的研究進展,對比了各類抑制方法的優缺點,并基于黃鐵礦的光電化學性質提出具有潛力的黃鐵礦抑制新方向。
1 調控黃鐵礦表面反應活性的意義
黃鐵礦晶胞及(100)晶面示意圖[7]如圖1 所示。黃鐵礦晶體呈立方體晶系,由S 原子和Fe 原子交替排布構成,晶格參數a=b=c=0.540 67 nm,晶格角α=β=γ=90°[6]。

圖1 黃鐵礦晶胞及(100)晶面示意圖[7]Fig. 1 Schematic diagram of pyrite cell and (100)surface[7]
在礦物加工過程中,浮選是分離黃鐵礦的最常見的工藝流程[11],而浮選藥劑是實現浮選分離的關鍵。在典型硫化礦中,天然可浮選能力從大到小依次為輝鉬礦、黃銅礦、方鉛礦、黃鐵礦、閃鋅礦,其中,黃鐵礦的天然可浮選處于中等。同時,不同產地或統一產地不同礦床的黃鐵礦天然可浮選能力存在較大差異[12]。在硫化礦浮選領域,最常用的捕收劑是黃藥類捕收劑(黃原酸鹽;ROCSS-),但是,除了(未經活化的)閃鋅礦和鐵閃鋅礦等外,黃藥類捕收劑可以同時捕收包括黃鐵礦在內的大部分硫化礦,難以實現有效分選(圖2)[13-17]。
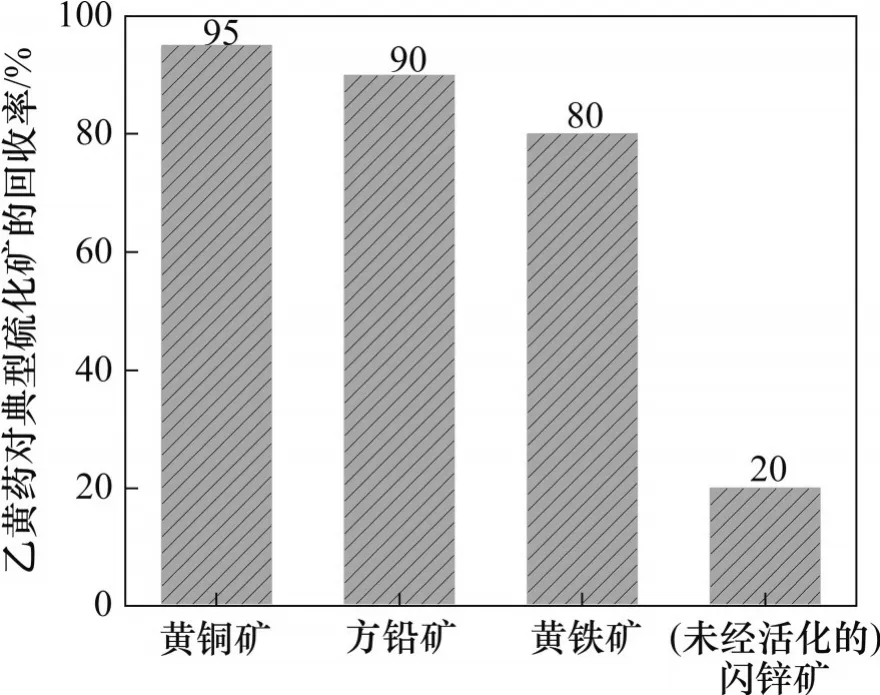
圖2 乙黃藥(EX)對硫化礦的回收率(EX濃度為2×10-4 mol/L)Fig. 2 Recoveries of sulfide minerals with EX(concentration of EX is 2×10-4 mol/L)
CHEN[18]論述了多種礦物與各類浮選藥劑的作用機理,發現浮選藥劑與礦物之間的作用主要分為正向σ鍵和反饋π鍵,前者由藥浮選藥劑提供孤對電子給礦物表面金屬離子的空軌道形成,后者由金屬離子提供π電子對與浮選藥劑的空π軌道作用形成。根據軟硬酸堿理論(HSAB)可知,硫化礦浮選以反饋π鍵為主[18]。圖3所示為黃鐵礦中八面體強場中Fe2+與黃藥(X-)的電子對-軌道相互作用模型和乙黃藥分子與黃鐵礦的吸附模型。黃藥提供孤對電子給Fe2+的eg空軌道,形成σ配位鍵,同時Fe2+的t2g軌道上的π 電子對與黃藥的空π 軌道作用,形成反饋π 鍵,作用過程中軌道對稱性匹配,且豎立吸附的黃藥分子極大地降低了吸附過程的空間位阻[18]。綜上可知,傳統的黃藥類捕收劑與黃鐵礦作用較強,難以實現黃鐵礦與其他有價礦物的高效分選。
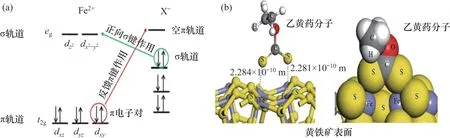
圖3 八面體強場中Fe2+與黃藥(X-)的電子對-軌道相互作用模型及乙黃藥與黃鐵礦的吸附模型[18]Fig. 3 Electron-orbital interaction of Fe2+ with a reagent molecule in a strong octahedral field and adsorption configuration of ethyl xanthate molecule on the surface of pyrite[18]
此外,黃藥類捕收劑在大部分硫化礦表面的吸附被認為是一種電化學過程,主要包括以下三個部分[4,19-21]:1) 礦物表面化學吸附黃原酸鹽離子(式(1));2) 礦物中金屬離子將黃原酸鹽氧化為二黃原酸鹽(式(2));3) 電荷轉移,形成金屬-黃原酸鹽(式(3)),浮選中的陽極氧化一般與氧的陰極還原反應同時進行(式(4))。
式中:X-、Xads、2X-、MS和MX2分別表示黃原酸鹽離子、被吸附的黃原酸鹽、二黃原酸鹽、硫化物礦物和金屬-黃原酸鹽;S 代表單質硫或多硫化物。
對于黃鐵礦而言,黃原酸鹽捕收劑在其表面的吸附主要包含以下4 個步驟[4,19-21]:1) 黃原酸鹽離子在黃鐵礦表面的吸附及鐵黃原酸鹽的形成;2) 黃原酸鹽離子氧化和二黃原酸鹽的形成;3) 二黃原酸鹽和黃原酸鹽在黃鐵礦表面通過物理作用、化學作用和前述表面化學反應產生共吸附;4) 被氣泡抓取實現上浮(圖4)[19,20,22]。硫化礦表面活性位點多為金屬陽離子(以M2+為主),上述金屬陽離子與傳統捕收劑的作用相似,這也是浮選藥劑選擇性差的根源。因此,靶向掩蓋或轉化黃鐵礦表面的活性位點,改變黃鐵礦表面Fe2+的賦存形態,進而降低黃鐵礦表面的反應活性,是實現黃鐵礦的有效抑制及與其他硫化礦高效分選的關鍵。
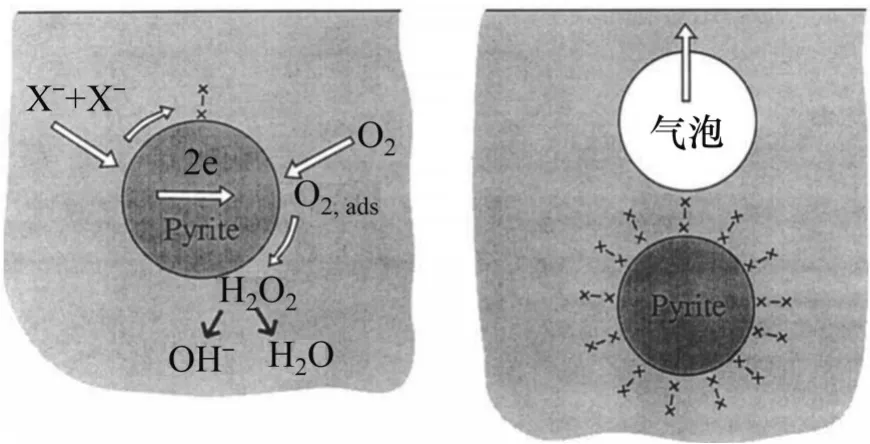
圖4 黃藥類捕收劑浮選黃鐵礦的經典模型示意圖[22]Fig. 4 Schematic presentation of classical model of pyrite flotation with xanthate collectors[22]
轉化活性位點的方法主要借助氧化劑或磨礦介質來實現。與新鮮黃鐵礦表面相比,黃藥在充分氧化的黃鐵礦表面上的吸附強度顯著降低,表明黃藥與二價鐵離子更親和,而不是FeOOH/Fe2O3等鐵的氫氧化物或氧化物。此外,深度氧化后的黃鐵礦表面潤濕性更強,這進一步限制了黃鐵礦的上浮[23]。Fe2O3、FeOOH 和FeS2中的Fe 3d 軌道及黃藥類捕收劑的—C=S 官能團的價電子構如圖5所示。FeOOH和Fe2O3中的Fe3+是d5高自旋離子,而FeS2中的Fe2+在d6電子組態中具有低自旋態;另外,黃藥S 原子的3p 軌道中有3d 空軌道和孤對電子[24-25]。鐵原子3d 軌道上的孤對電子對或空軌道的存在是黃藥S原子與鐵原子配位的先決條件。然而,在FeOOH和Fe2O3的d5高自旋Fe3+離子中既不存在孤對電子對,也不存在空軌道,當FeOOH 和Fe2O3的Fe 原子與黃藥S 原子配位時,它們的電子構型不支持σ鍵或π鍵的形成[23]。本質上,黃藥在FeOOH/Fe2O3和FeS2表面的選擇性吸附是它們的Fe原子的電子構型不同造成的[23]。
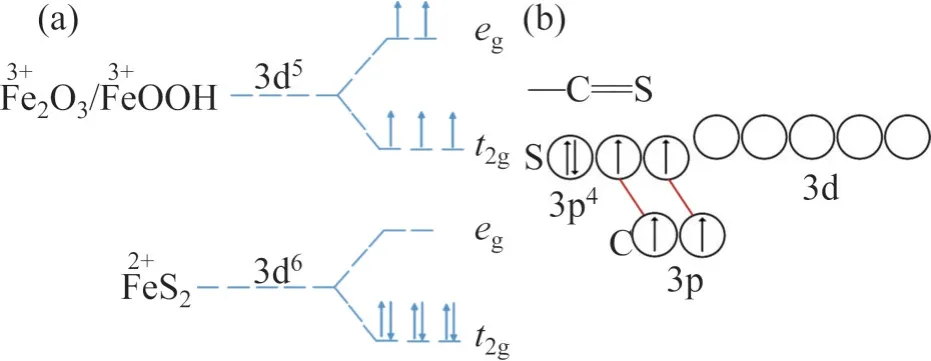
圖5 Fe2O3,FeOOH和FeS2中的Fe 3d軌道及黃藥類捕收劑的—C=S官能團的價電子構型[23]Fig. 5 Valence electron configurations of Fe 3d orbitals of Fe2O3, FeOOH and FeS2 and —C=S functional group of xanthate collectors[23]
2 調控黃鐵礦表面反應活性的主要途徑
2.1 有機抑制法
現階段有機抑制法主要通過有機抑制劑的化學吸附(配位作用為主)和物理吸附(氫鍵、范德華力等)實現,吸附在黃鐵礦表面的有機抑制劑會掩蓋其活性位點,從而達到抑制效果。有機抑制劑按相對分子質量可分為大分子有機抑制劑和小分子有機抑制劑。大分子有機抑制劑種類繁多,如聚丙烯酰胺、改性木質素磺酸鹽生物聚合物、淀粉、CMC、糊精、海藻酸鈉等[4,26-27]。其中,多糖類抑制劑是大分子有機抑制劑中最常見的一類,大多來自自然界中的天然物質,如淀粉、瓜爾豆膠和殼聚糖等,具有生物可降解、無毒無害的優點。有機抑制劑的分子結構中通常含有—COO-、—、—NH2、—CSS-等多種官能團,上述官能團中的雜原子(N、O、S、P)通過與黃鐵礦表面的鐵離子或鐵的氫氧化物及氧化物產生螯合作用實現對黃鐵礦表面的匹配和吸附[4,28-30],掩蓋其活性位點,同時結構中—OH等親水官能團可以有效增加黃鐵礦表面的親水性,協同阻止黃鐵礦的上浮。
硫化礦小分子有機抑制劑同樣依賴于由雜原子或雜環結構構建的親固端[31-33],小分子有機抑制劑的整體抑制效果通常比大分子有機抑制劑的抑制效果弱。王瑜等[33]探究了巰基乙酸和巰基乙醇對黃鐵礦的抑制作用并對抑制過程進行了密度泛函理論(density functional theory;DFT)計算。結果表明,黃鐵礦{100}晶面具有更多的表面活性位點,巰基類小分子抑制劑更易于在黃鐵礦{100}晶面吸附,進而占據和掩蓋其表面活性位點,實現對黃鐵礦的有效抑制(圖6)。熊道陵等[34]開發了一種新型黃鐵礦有機抑制劑三羧基甲基—二硫代碳酸鈉,在pH=9~12的礦漿中,2.4 mmol/L的三羧基甲基—二硫代碳酸鈉對黃鐵礦抑制效果較好,對黃銅礦抑制作用較弱,可以實現低堿條件下的銅硫分離,DFT 計算結果表明,相比黃銅礦,三羧基甲基—二硫代碳酸鈉對黃鐵礦表面的吸附作用更強。
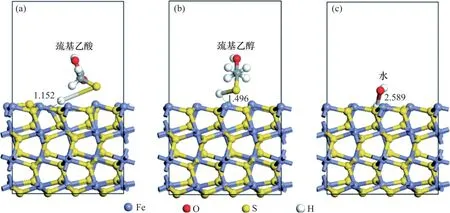
圖6 巰基乙酸和巰基乙醇對黃鐵礦抑制作用的DFT計算[33]Fig. 6 DFT calculation of the depression effect of mercaptoacetic acid and mercaptoethanol on pyrite[33]
相比上述小分子有機抑制劑,大分子有機抑制劑含有更多的螯合官能團(—COO-、—、—OH、—NH2、—CSS-等),對黃鐵礦表面的吸附作用更強,因而具有更優異的抑制效果[35]。BOULTON等[36]的研究表明,不同取代基的聚丙烯酰胺聚合物(PAM)均可抑制黃鐵礦,抑制能力由強到弱依次為羥基PAM、羰基PAM、硫脲PAM、磺基PAM。上述不同取代基的PAM 在鋅-鐵硫化礦分離體系中,對黃鐵礦的選擇性抑制能力由強到弱(即抑制后閃鋅礦和黃鐵礦的回收率差值由大到小)依次為羰基PAM、磺基PAM、羥基PAM、硫脲基PAM(圖7)。造成上述現象的原因可能是:不同取代基親水能力存在差異;不同取代基與黃鐵礦表面金屬離子的軌道匹配程度存在差異;吸附不同取代基的晶體場穩定化能存在差異;不同取代基的極性及電性存在差異,綜合導致不同取代基的PAM與黃鐵礦的作用強度和作用選擇性存在差異。PAM 抑制黃鐵礦的主要機理同樣是通過吸附作用掩蓋黃鐵礦表面的活性位點,同時增強其潤濕性。LóPEZ VALDIVIESO 等[37]的研究結果表明低成本且無毒無害的糊精同樣是黃鐵礦的高效抑制劑。除了吸附作用外,糊精結構中的眾多羥基官能團顯著增加了黃鐵礦表面的親水性(圖8)。

圖7 PAM及取代基-PAM的化學結構[36]Fig. 7 Chemical structures of the PAM and substituted-PAM[36]

圖8 糊精在黃鐵礦表面的吸附構型[37]Fig. 8 Adsorption configuration of dextrin on pyrite surface[37]
有機抑制劑的抑制能力與自身的結構(官能團種類、數量等)和性質(相對分子質量、電負性和溶解度等)密切相關[38-40]。馮博等[41]研究表明,羧甲基纖維素(CMC)能夠通過靜電作用吸附在黃鐵礦表面影響其浮選,CMC的分子量越高,其抑制效果越明顯。BICAK等[42]研究表明,瓜爾豆膠在0.1×10-6的低用量下就可以有效吸附在黃鐵礦表面達到抑制效果,而CMC則需要更大的藥劑用量才能實現對黃鐵礦的有效抑制,這主要是由于CMC中帶高負電荷的取代基(—COO-、—CH2O-、—CH2COO-)與表面同樣帶負電荷的黃鐵礦之間存在靜電排斥作用。在CMC中,羧甲基和羥基都與礦物表面的金屬位點相互作用。羧甲基與金屬陽離子和金屬羥基相互作用,羥基與金屬羥基相互作用,其相互作用強度取決于pH,在表面金屬離子完全羥基化的pH下,相互作用最強[42]。
通過調控大分子有機抑制劑的相對分子質量和藥劑制度,以及與其他浮選藥劑聯用,可使其抑制選擇性大幅提高。例如,殼聚糖及其衍生物可以吸附在絕大多數的硫化礦表面,即同時抑制多種硫化礦,造成目的礦物的損失及精礦品位的降低。研究表明[43],殼聚糖的抑制效果和它的相對分子質量和用量密切相關,高相對分子質量(500~1 000 kDa)的殼聚糖在低用量時便可抑制多種硫化礦,低相對分子質量(1~50 kDa)的殼聚糖(寡糖)在低用量時抑制效果不佳,中等相對分子質量(50~500 kDa)的殼聚糖在低用量時具有更優的抑制選擇性。同時,FENG等[44]的研究表明,加藥順序對羧甲基纖維素鈉(CMC)對黃鐵礦的抑制效果有明顯影響,在戊基黃原酸鈉(PAX)預吸附層的存在下,由于競爭性吸附,CMC 對黃鐵礦的抑制作用減弱。由此可見,大分子有機抑制劑具有選擇性抑制黃鐵礦的能力,但與其相對分子質量、藥劑制度、加藥順序及礦漿pH等諸多因素密切相關。
2.2 微生物抑制法
微生物抑制劑的抑制過程溫和環保,本身具有無毒無害無污染的優點,符合綠色礦山的理念,具有良好的發展前景。氧化亞鐵硫桿菌是最常用的黃鐵礦抑制菌落,抑制效果顯著,其抑制黃鐵礦活性位點的途徑包括吸附掩蓋和氧化轉化兩種。在作用時間較短時,主要通過吸附掩蓋黃鐵礦表面的活性位點實現抑制;隨著作用時間的增加,黃鐵礦表面活性位點的氧化轉化成為抑制的主要途徑[45-46]。CHANDRAPRABHA 等[47]發現氧化亞鐵硫桿菌可以用于從黃鐵礦和黃銅礦混合物中選擇性地抑制分離黃鐵礦。MARTíN 等[48-49]的研究也表明氧化亞鐵酸硫桿菌可以作為黃鐵礦的有效抑制劑。該研究在淡水、鹽水(35 g/L NaCl,與海水中的鹽濃度相對應)和海水三個系統中研究了氧化亞鐵酸硫桿菌對黃鐵礦的抑制結果。結果表明,在淡水、鹽水與海水中,用氧化亞鐵酸硫桿菌處理黃鐵礦后,黃鐵礦在pH=8 時的回收率分別從99%下降到24%、34%和36%。
但是由于氧化亞鐵硫桿菌屬于化能自養菌,培養過程耗能高且生長緩慢,菌體得率較低。同時這種菌體多在酸性條件下使用,使用時會伴隨大量選礦酸性廢水的排放,給后續廢水處理帶來困難,其工業化應用受到一定限制[45-46]。因此,其他微生物抑制劑的篩選和開發引起眾多研究人員的關注。BLEEZE等[50]研究發現氧化亞鐵鉤端螺旋菌對黃鐵礦和黃銅礦混合物中的黃鐵礦有選擇性吸附及抑制的現象。不同處理時間時黃鐵礦和黃銅礦表面的SEM 圖像如圖9 和圖10 所示。從圖9和圖10 可以看出,大量氧化亞鐵鉤端螺旋菌吸附在黃鐵礦表面,而其在黃銅礦表面的吸附并不明顯。在處理時間為0~168 h 時,該現象一直存在;在處理時間為48 h 時,黃銅礦中黃鐵礦的選擇性抑制和分離效果最佳。

圖9 不同處理時間時黃鐵礦表面的SEM圖像[50]Fig. 9 SEM images of pyrite surface in different processing time[50]

圖10 不同處理時間時時黃銅礦表面的SEM圖像[50]Fig. 10 SEM images of chalcopyrite surface in different processing time[50]
張興等[51]比較了親水性較好的3 種細菌(氧化亞鐵硫桿菌、球形紅假單胞菌和氧化硫硫桿菌)對黃鐵礦的抑制作用,結果表明,這三種細菌作用5 min便可以將黃鐵礦有效抑制,使黃鐵礦的回收率分別下降至為7.66%、8.92%和10.29%。因為抑制時間僅為5 min,所以此時吸附掩蓋黃鐵礦表面的活性位點是這三種細菌抑制黃鐵礦的主要途徑。同時,該研究發現細菌與黃鐵礦的吸附與脫吸附處于動態變化過程。張明旭等[52]的研究進一步證實了球形紅假單胞菌在實際礦的浮選中,該細菌對黃鐵礦仍保持著良好的抑制效果。梁海軍等[53]的研究表明,菌體抑制是通過減少浮選捕收劑在黃鐵礦表面的吸附而實現,靜電作用和空間位阻效應是導致黃鐵礦有效抑制的重要原因。因此,親水性的菌體不僅通過吸附掩蓋了黃鐵礦表面原本用于結合捕收劑的活性位點,黏附在黃鐵礦表面的菌體本身也會對捕收劑的吸附造成阻礙,進一步降低黃鐵礦表面與捕收劑分子親和的可能性,達到抑制黃鐵礦的目的。
微生物抑制劑除了可以通過吸附掩蓋黃鐵礦表面的活性位點來抑制黃鐵礦,也可以通過氧化轉化活性位點實現對黃鐵礦的有效抑制。利用微生物代替傳統無機抑制劑氧化黃鐵礦表面,顯著降低了選礦過程對周邊環境的影響。同時,微生物氧化時會針對特定礦物培養特定菌落,針對性更強,抑制的選擇性更為優異。沃克德等[54]的研究證明了在酸性條件下,多種氧化亞鐵硫桿菌菌株(嗜酸)均可以氧化黃鐵礦,以達到抑制黃鐵礦的目的(表1)。MEHRABANI 等[46,55]的研究也證實了氧化亞鐵硫桿菌可以作為一種無毒抑制劑在黃鐵礦和閃鋅礦的選擇性浮選過程中替代劇毒有機抑制劑NaCN,氧化亞鐵硫桿菌抑制黃鐵礦的最佳用量為4×106個/mL。GU 等[56]研究了氧化亞鐵硫桿菌對黃鐵礦氧化速率的影響,實驗結果表明氧化亞鐵硫桿菌的存在增大了極化電流密度,加速了黃鐵礦的氧化速率。根據過往研究,微生物通過氧化抑制黃鐵礦的電化學反應原理如下:
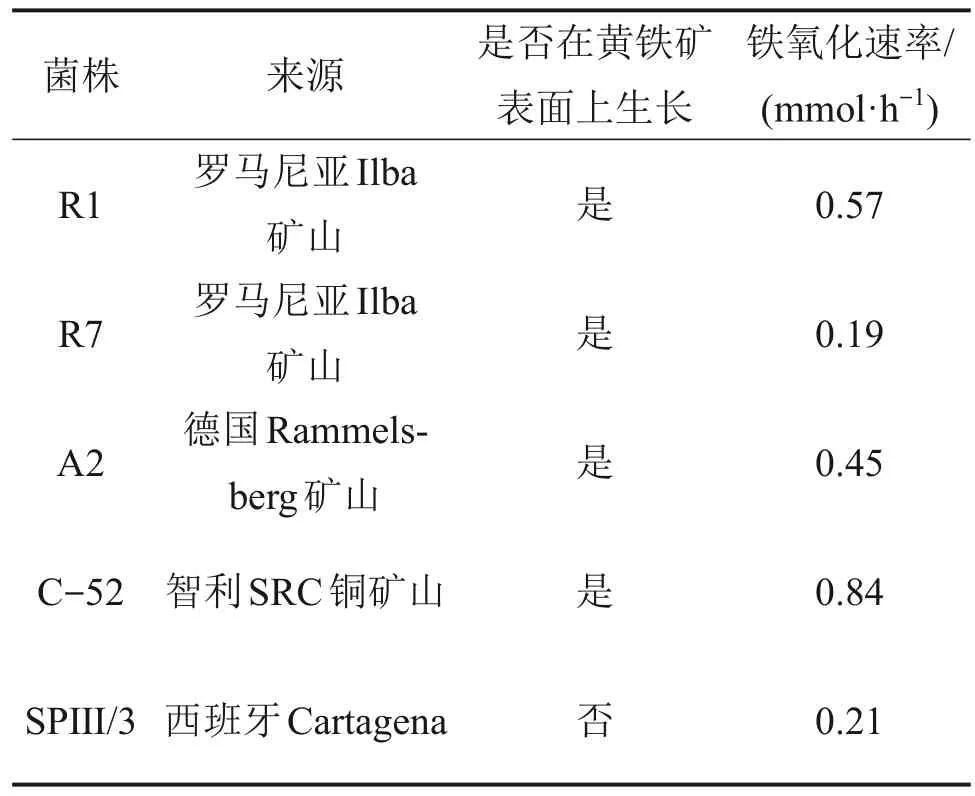
表1 氧化亞鐵硫桿菌菌株及其對黃鐵礦的抑制效果[54]Table 1 Thiobacillus ferrooxidans and their depression effect on pyrite[54]
2.3 磨礦抑制法
硫化礦的表面性質和礦漿環境的性質(pH、電位、電導率、溶解氧含量和離子種類及濃度)決定了硫化礦的可浮性(圖11 和圖12)[57],而磨礦過程會在很大程度上影響硫化礦的表面性質[58]。在磨礦過程中,硫化礦和磨礦介質之間以及硫化礦各組分之間接觸時會發生電化學反應,由于硫化礦各組分及磨礦介質材料的靜電位存在差異,產生了接觸電流,從而在硫化礦與磨礦介質之間以及硫化礦之間發生電化學反應,靜電位高的一端充當陰極,發生還原反應,靜電位低的一端充當陽極,發生氧化反應[59]。上述電化學反應會在硫化礦的表面生成新的產物從而改變硫化礦的表面性質,進而影響硫化礦后續的浮選行為和分離效果[59]。
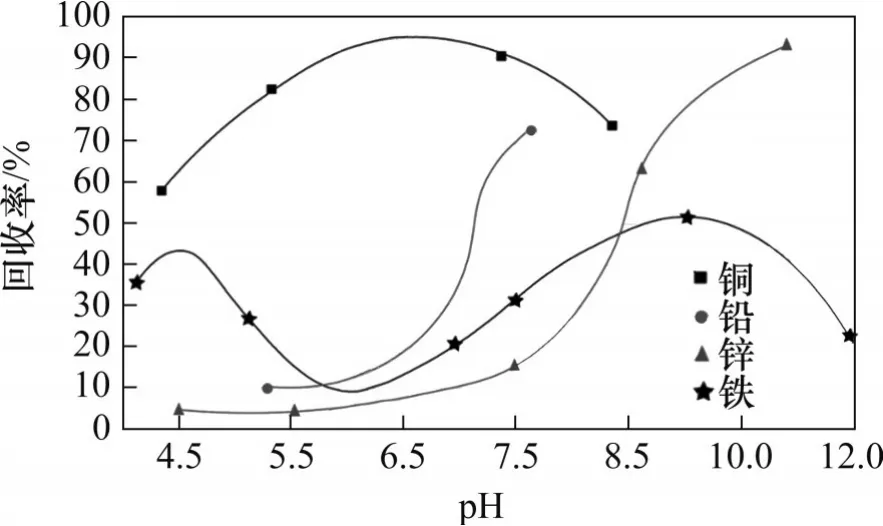
圖11 銅/鉛/鋅/鐵硫化礦的回收率與礦漿pH的關系[57]Fig. 11 Recoveries of Cu/Pb/Zn/Fe sulfide minerals versus pulp pH[57]

圖12 硫化礦的浮選行為與Eh/pH的關系[57]Fig. 12 Flotation behaviors of sulfide minerals versus Eh/pH[57]
中南大學顧幗華教授課題組[60-62]、北京礦冶研究總院的何發鈺研究員課題組[63]以及北京科技大學孫體昌課題組[64]的研究表明,在鐵罐-鐵球體系下進行磨礦,會產生較低的礦漿電位和更強的還原氛圍。同時,黃鐵礦在用介質磨礦時電位下降幅度明顯,這是由于體系中鐵離子(來自于磨礦介質)的存在,當兩者相互接觸時會形成Galvanic 電偶及氧化還原反應(圖13 和圖14)[65-66],此時磨礦體系中會存在如下平衡關系:
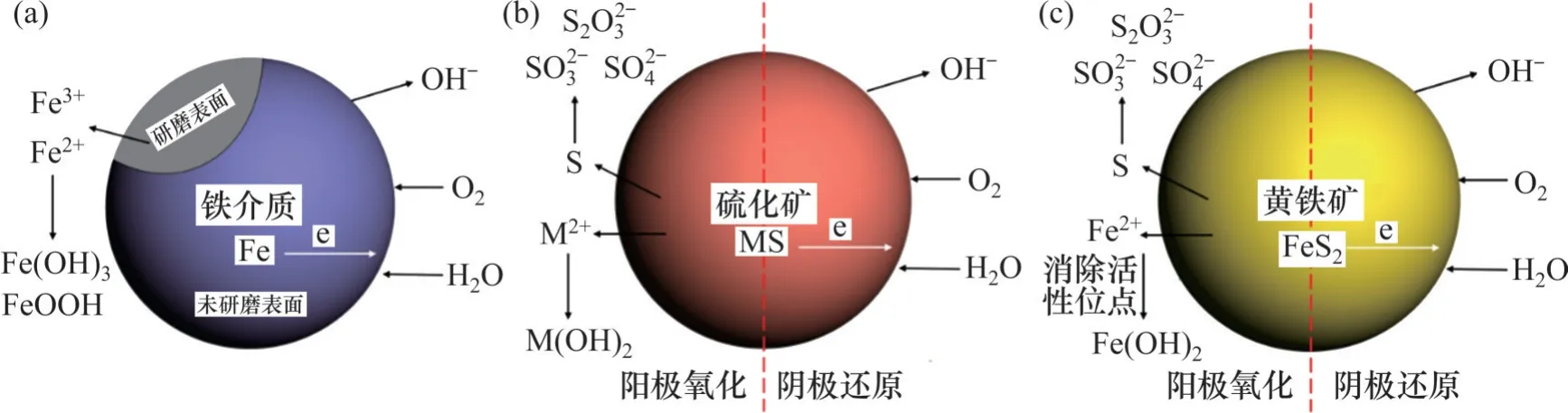
圖13 自身局部電池和自氧化過程Fig. 13 Local battery and autoxidation process
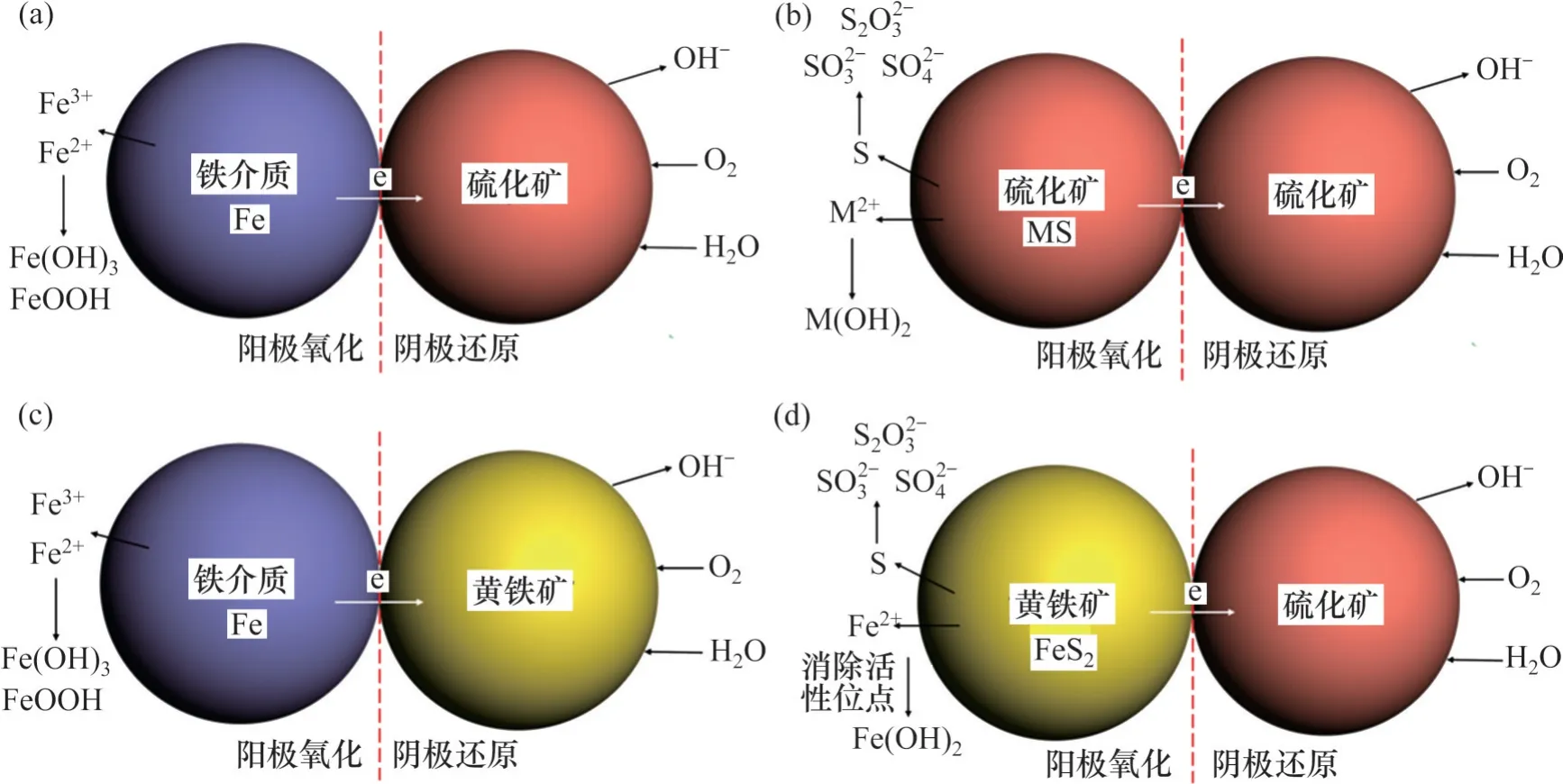
圖14 Galvanic電偶及氧化還原反應Fig. 14 Galvanic couple and redox reactions
由這些反應式可看出,黃鐵礦表面活性位點Fe2+經過磨礦氧化后會產生鐵的氫氧化物,這些產物形成時會覆蓋在黃鐵礦表面,阻止捕收劑的吸附,使黃鐵礦的回收率下降。黃鐵礦的回收率會隨礦漿pH的增加而降低,這是因為pH越大OH-濃度越高,形成的氫氧化物沉淀也會越多,從而降低了黃鐵礦的回收率。黃鐵礦表面氧化程度由強到弱的順序為濕式鐵磨、濕式瓷磨(干式鐵磨)、干式瓷磨,除濕式鐵介質磨礦外,其他三種磨礦方式下均產生了缺金屬富硫表面。黃鐵礦經磨礦后的可浮性由強到弱的順序為干式瓷磨、干式鐵磨、濕式瓷磨、濕式鐵磨[60-62]。鐵介質磨礦后的黃鐵礦表面粗糙,腐蝕嚴重,存在大量的非晶質化親水性的FeOOH,這是造成其表面親水性增強、浮選回收率低的主要原因[63,67]。
在黃鐵礦-方鉛礦浮選體系中,磨礦對黃鐵礦的選擇性抑制作用較強,這是因為相比方鉛礦,黃鐵礦的靜電位更高,更容易被氧化[17]。PENG等[68]的研究表明:磨礦環境對方鉛礦浮選及黃鐵礦中方鉛礦的分離有顯著影響,這種效應與礦物表面存在的鉛和鐵的氧化態密切相關。鐵氧化態對方鉛礦和黃鐵礦浮選均有抑制作用,而鉛氧化態對黃鐵礦浮選作用不大。通過選擇能夠控制鉛和鐵氧化的磨礦條件,可獲得最佳方鉛礦浮選和方鉛礦對黃鐵礦的選擇性(圖15)。在黃鐵礦-黃銅礦浮選體系中,磨礦介質與分選效果同樣緊密相關。PENG等[69]的研究表明,黃銅礦與黃鐵礦混合后,磨礦介質中鐵氧化物質的含量增加,會導致黃銅礦浮選回收率降低。由于被Cu2+活化,黃鐵礦回收率明顯提高。因此,當采用磨礦氧化黃鐵礦時,要注意磨礦體系中應避免銅礦物或含銅介質[70],控制磨礦時間和細度,減少微細粒礦物溶解產生,以免對后續浮選分離造成不利影響[71]。
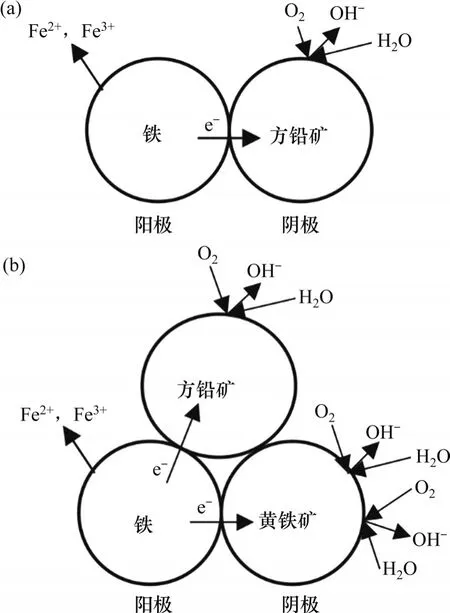
圖15 黃鐵礦-方鉛礦分選體系中的電流相互作用[68]Fig. 15 Interaction of electric current in pyrite-galena separation system[68]
除上述原因外,由于黃鐵礦富含Fe2+和Fe3+,易于發生芬頓(Fenton)作用,因此黃鐵礦磨礦過程會與水分子或氧氣反應產生羥基自由基、超氧陰離子自由基,并進一步產生過氧化氫,增加黃鐵礦的氧化程度,從而降低了其可浮性[72]。除了借助單一磨礦過程氧化黃鐵礦,在磨礦過程中添加浮選藥劑也可以有效提升黃鐵礦選擇性抑制效果。基于磨礦過程的黃鐵礦電位調控浮選抑制工藝已被成功應用于工業實踐。以凡口鉛鋅礦為例,選礦廠技術人員與高校研究人員的聯合研究[73-74]表明,電位調控浮選工藝成功應用于凡口鉛鋅礦磨礦過程,顯著提高了該選礦廠的生產技術指標。具體方案為:將石灰(調整劑/抑制劑)與乙硫氮(捕收劑)同時加入磨機,充分利用石灰對礦漿電位的調控與穩定作用以及在低氧化電位下乙硫氮對礦物的選擇作用實現鉛-鋅-鐵的高效分選。
2.4 無機抑制法
氰化物是黃鐵礦的傳統無機抑制劑,抑制效果顯著,但氰化物是最強烈、毒性最高的化合物之一,對環境的影響極大,近年來逐漸被其他抑制劑替代[75-80]。氰化物對黃鐵礦浮選的抑制作用可能是由以下一種或多種機制引起的[81-84]:1) 氰化物對黃鐵礦表面黃原酸鹽離子的替代作用,類似于氫氧化物對黃鐵礦浮選的抑制作用;2) 氰化物在黃鐵礦表面的吸附導致不溶性氰化鐵化合物的形成,這些不溶性氰化鐵化合物在黃鐵礦表面預富集,使黃鐵礦表面親水,抑制了黃原酸鹽離子的吸附;3) 氰化物也可以與表面氧化產生的硫或多硫化物反應形成硫氰酸鹽,硫氰酸鹽是親水的,被吸附后抑制了黃鐵礦;4) 氰化物的加入可以降低混合電位,抑制黃鐵礦表面的電化學活性,干擾黃原酸鹽的化學吸附和氧化。
GUO等[81-83]研究表明,在氰化物(例如游離氰化物或氰化亞銅等)存在的情況下,黃原酸溶液仍有可能被氧化,雙黃原酸不能吸附在黃鐵礦上。黃鐵礦表面活性位點的“毒化”是由于黃鐵礦表面形成了氰化鐵化合物。氰化鐵化合物主要以的形式優先吸附在黃鐵礦表面,起到阻礙黃鐵礦與其他礦物表面反應的動力屏障作用,從而抑制了黃鐵礦浮選。此外,氰化物還能去除黃鐵礦表面的富硫疏水物質,進一步降低黃鐵礦的可浮性。在-200 mV 到200 mV 的電位范圍內,0.1 mmol的NaCN可以顯著抑制黃原酸在黃鐵礦上的化學吸附和氧化,使黃鐵礦表面疏水。ZHAO等[85]從實驗和計算模擬的角度研究了氰化物離子對黃鐵礦、白鐵礦和磁黃鐵礦的抑制作用,結果表明氰化物離子對這三種硫化礦均有明顯的抑制作用,其中對白鐵礦的抑制作用最強,其次是磁黃鐵礦和黃鐵礦。
除上述原因外,研究表明,氰化物的吸附作用以反饋π 鍵作用為主,而黃鐵礦能夠提供3 對π電子,容易被氰化物抑制,并且添加氰化物可以使黃鐵礦開始氧化的陽極電位產生明顯負移,即氰化物在黃鐵礦表面的吸附促進了Fe2+的氧化,使其π 電子從3 對減少為2 對,減弱了黃藥等捕收劑在黃鐵礦表面的吸附作用,進一步提升了抑制效果[18]。
通過添加石灰調節礦漿為高鈣、高堿環境是抑制黃鐵礦表面活性的常見手段,這是由于相較黃銅礦、方鉛礦和(銅活化)閃鋅礦等有機硫化礦,黃鐵礦的浮選臨界pH較低,例如,在選用乙黃藥作為捕收劑且用量為25 mg/L 時,黃銅礦、方鉛礦、(銅活化)閃鋅礦和黃鐵礦的浮選臨界pH 分別為11.8、11.5、13.3 和10.5[18,86]。相比傳統的礦漿pH 調整劑氫氧化鈉,在含黃鐵礦的浮選中,石灰的應用更為廣泛且抑制效果更好。石灰對黃鐵礦更為優異的抑制作用主要是因為[18]:羥基鈣有空π軌道,而氫氧根離子沒有空π軌道,羥基鈣為強場配體,氫氧根離子為弱場配體,這意味著在黃鐵礦表面,石灰比氫氧化鈉的作用更強、更穩定。這一結論也被晶體場理論(CFT)證實:羥基鈣和氫氧根離子在黃鐵礦表面吸附后,晶體場穩定化能(CFSE)分別為-24 Dq 和-4 Dq,更負的CFSE 意味著更穩定的配位結構。石灰是當前抑制黃鐵礦的最常用的無機抑制劑之一,在工業上應用十分廣泛,但大量石灰的使用會造成管道結垢,并增加后續廢水的處理難度[87-89]。
除了氰化物(CN-)和石灰(CaOH+)外,黃鐵礦也易于被亞硫酸鈉抑制,而硫化鈉/硫氫化鈉(HS-)和氫氧化鈉(OH-)對黃鐵礦的抑制效果欠佳,這是因為亞硫酸根、氰化物及羥基鈣都可以在與黃鐵礦表面Fe2+吸附過程中形成反饋π鍵;而硫氫根離子作用過程中只有微弱的反饋π 鍵作用,主要以σ鍵作用為主;氫氧根離子作用過程中只有σ鍵作用,常見抑制劑的分子軌道如圖16所示[18]。
3 總結與展望
現有抑制黃鐵礦的方法大多是通過改變黃鐵礦表面性質或掩蓋黃鐵礦表面吸附位點來實現的。現有的抑制方法對黃鐵礦抑制效果良好且各有優勢。借助微生物和大分子有機抑制劑的黃鐵礦抑制方法更為環保,更符合綠色礦山的理念;借助無機抑制劑的黃鐵礦抑制方法的成本低、效果顯著,適用于規模工業化生產;借助磨礦(電位調控浮選)和小分子有機抑制劑的黃鐵礦抑制方法也相對環保,并且選擇性較好。但上述方法仍存在一定的不足,例如,已報道的微生物法主要采用氧化亞鐵硫桿菌,菌種單一,生長緩慢,菌體得率低,工業化應用受到限制;現有的磨礦抑制法對低品位、嵌布粒度細的復雜硫化礦處理效果差;無機抑制劑中氰化物的毒性很大且污染嚴重,使用石灰等的高堿抑制法會導致管道結垢并造成選礦廢水處理難度增大;大分子有機抑制劑的抑制選擇性較差,在抑制黃鐵礦的同時會抑制其他有用礦物,造成有用礦物的損失;小分子抑制劑的成本較高,不利于工業應用。因此,就吸附掩蓋活性位點和氧化轉化活性位點兩個方向發展綠色、環保、高效、經濟的黃鐵礦選擇性抑制新方法是至關重要的。
在吸附掩蓋活性位點方面,在中性環保的條件下實現黃鐵礦的靶向抑制主要依賴高效、高選擇性的新型抑制劑的開發。浮選藥劑的開發經歷了三個階段:第一階段,經驗指導實踐;第二階段,理論指導實踐;第三階段,數據指導實踐。毫無疑問,第一階段中,基于經驗和試錯法的捕收劑開發方法耗時耗力。第二階段中,理論計算快速發展,如吸附能和吸附構型的計算,促進了對表面活性劑浮選性能和分離機理的進一步探索。然而,在高通量處理浮選藥劑時,大多數周期性計算都是昂貴且耗時的。在第三階段,人們希望可以依靠(定量)構效關系(SAR/QSAR)和機器學習(ML)等人工智能(AI)技術,通過大數據分析實現對抑制劑等浮選藥劑的高通量和定量預測。當前該類方法主要用于預測具有相同骨架的浮選藥劑,準確性高但應用范圍較窄,實現人工智能浮選(AIFlotation)仍道阻且長。在未來研究中,可以借助真實的浮選數據,綜合考慮浮選藥劑參數(電性、極性等)、浮選參數(藥劑用量、礦漿pH、浮選時間等)、晶面參數(電性、晶胞參數等)和晶面與浮選藥劑及水分子的作用(軌道匹配、空間位阻、晶體場穩定化能等)參數進行特征工程研究,開發高通量篩選浮選藥劑的新技術框架與新方法,實現高通量篩選,預測各類浮選藥劑分子的浮選行為,且不再受限于特定骨架結構,為浮選藥劑的開發提供通用的方法和技術支持。同時需要注意的是,在人工智能浮選探索的過程中,在算法改進的同時仍需通過SAR/QSAR方法促進相關理論的進步。
在氧化轉化活性位點方面,大多數金屬硫化物是半導體,硫化物礦物浮選系統中會發生各種電化學反應。大量的研究表明,黃鐵礦浮選與電化學反應具有很強的相關性。黃鐵礦表面氧化程度的微小變化會顯著影響其可浮性。黃鐵礦被氧化后常常會形成親水和穩定的金屬氧化物/氫氧化物以降低可浮性,達到抑制效果。因而,通過氧化抑制黃鐵礦是最直接也是最根本的方法,但是現有常規氧化劑存在用量大、不環保及廢水難處理等缺點。作為礦物半導體,黃鐵礦可以在太陽輻射下吸收和轉換光子以產生電子,是制造低成本光伏太陽能電池板的一種豐富、無毒、廉價的原材料。與其他硫化礦相比,黃鐵礦具有適當的禁帶寬度(Eg=0.9 eV)與較高的光吸收系數(105 cm-1),其衍生材料已被作為光催化劑用于降解有機污染物或重金屬污染物。同時,黃鐵礦(FeS2)富含Fe2+,具備參與光催化氧化過程的可能性。基于上述觀點,本文作者認為未來可以基于黃鐵礦與其他硫化礦氧化還原電位的差異,結合經濟、可再生、清潔和安全的光電催化技術,選擇合適的光催化劑或光敏化劑靶向鈍化黃鐵礦表面,實現黃鐵礦的選擇性氧化抑制。
