兒童罕見病藥物治療新進展
2023-07-17 07:17:38李佳琦綜述王慧君周文浩審校
中國當代兒科雜志 2023年7期
李佳琦 綜述 王慧君 周文浩 審校
(復旦大學附屬兒科醫院新生兒科,上海 201102)
1983 年,美國《孤兒藥物法》首次提出罕見病的概念[1]。中國將罕見病定義為新生兒發病率小于1∶10 000或患病率小于1∶10 000,或總的患病人數小于14 萬的疾病[2]。截至2022 年,美國食品和藥物管理局(Food and Drug Administration,FDA)推動的“孤兒產品臨床試驗資助計劃”已使80 多種孤兒藥獲得FDA 批準[3]。目前,我國孤兒藥的本土化生產也日益受到重視[4]。與成人相比,兒童罕見病更常見,且危害更大,常危及生命。本文圍繞兒童罕見病藥物治療新進展進行綜述,為我國兒童罕見病研究與治療提供參考。我們將按酶替代療法(enzyme replacement therapy,ERT)和重組因子、小分子藥物、單克隆抗體、基因療法、超適應證藥物等5類罕見病藥進行逐一綜述。
1 ERT和重組因子
ERT 是一種通過重組DNA 技術合成重組酶,將其遞送至體內替代缺失或功能不全的酶的方法[5],常用于治療溶酶體貯積癥(lysosomal storage diseases,LSDs)。
溶酶體是一種負責降解細胞內各種內外源性物質的細胞器,編碼各種溶酶體酶的基因的缺陷會引起LSDs。LSDs 是50 余種溶酶體代謝病的統稱,總患病率為1∶7 700[6]。LSDs 主要為常染色體隱性(autosomal recessive,AR)遺傳模式致病,以黏多糖貯積癥(mucopolysaccharidoses,MPS)較常見,MPS 包括MPS Ⅰ、MPS Ⅱ、MPS ⅢA 等11 型[7]。MPS 主要呈AR 遺傳,而MPS Ⅱ和Fabry病為X 連鎖隱性遺傳,Danon 病為X 連鎖顯性遺傳。
LSDs 中,溶酶體酶的缺失主要導致糖胺聚糖的堆積,對骨骼、結締組織、神經系統等器官系統造成損害。MPS Ⅰ由編碼α-L-艾杜糖醛酸酶的IDUA基因突變引起[8];MPSⅡ由編碼艾杜糖醛酸-2-硫酸酯酶的IDS基因突變引起[9];MPS ⅢA 由SGSH基因突變使heparan-N-sulfamidase 缺乏引起[10];MPS ⅣA 由GALNS基因突變導致N-乙酰半乳糖胺-6-硫酸酯酶缺乏引起[11];MPS Ⅶ由編碼β-葡糖苷酸酶的GUSB基因突變引起[12];Fabry 病由GLA基因突變引起[13];尼曼-皮克病由編碼酸性鞘磷脂酶的SMPD1基因突變引起[14];戈謝病由GBA1基因突變引起,使得β-葡糖腦苷脂酶的活性降低[15]。ERT可有效增加上述疾病酶濃度(表1),早期獲得診斷并及時干預可有效延緩此類患兒的疾病進程。
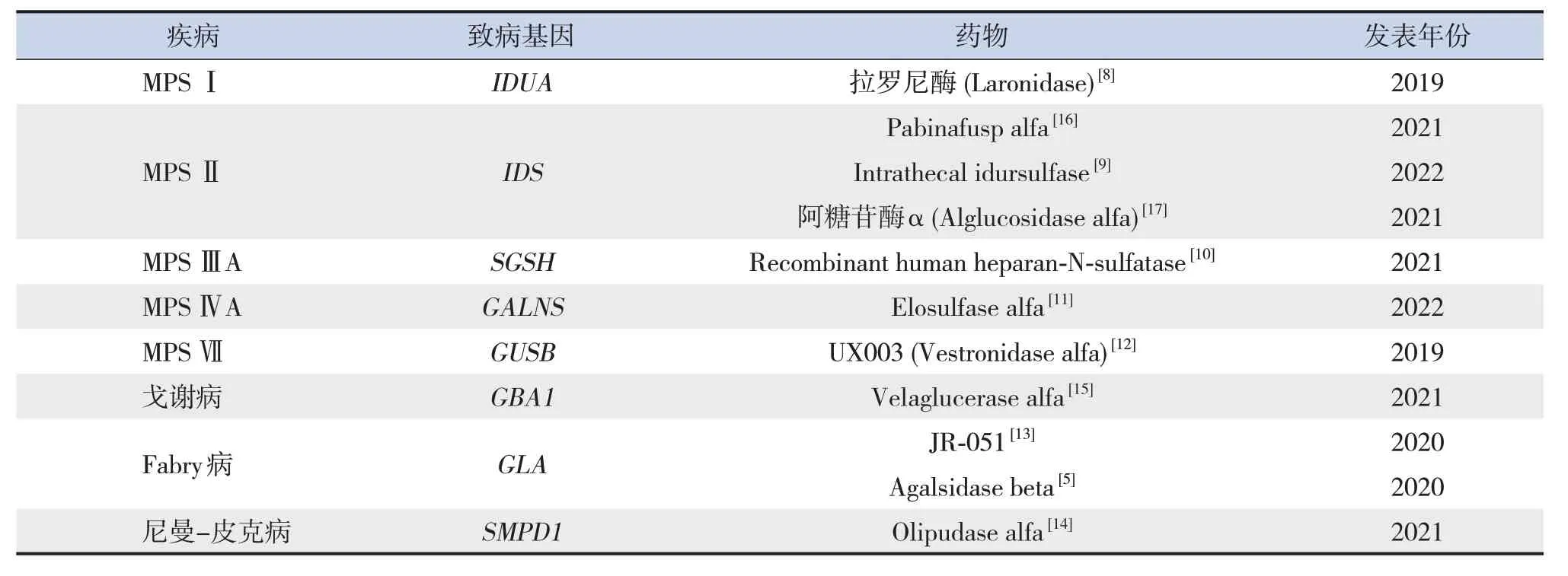
表1 酶替代療法治療溶酶體貯積癥總結
Pabinafusp alfa 可穿過血腦屏障,治療MPSⅡ[16]。但是,大多數ERT 藥物仍難以穿過血腦屏障,且存在靜脈注射產生抗藥物抗體的風險[7]。
2 小分子藥物治療
小分子藥物的優勢是能夠到達包括中樞神經系統在內的所有組織,并且成本低、可口服,易于制作[18],占上市罕見病藥物的80%~90%[19]。
2.1 囊性纖維化
囊性纖維化(cystic fibrosis, CF, MIM:219700)是一種AR遺傳的進行性多系統疾病,患病率為1∶6 000~1∶3 000,新生兒期即可起病[20]。CF 因囊性纖維化跨膜傳導調節因子(cystic fibrosis transmembrane conductance regulator,CFTR)的門控突變導致CFTR 蛋白異常,引起細胞膜對氯化物的運輸障礙[21],最常見的門控突變體是G551D[22]。
第一個被用于治療CF 的是CFTR 增效劑Ivacaftor,該藥主要通過增加G551D-CFTR 氯離子通道開放以增強CFTR 蛋白功能[23]。2022 年,Liu等[24]研發出一種新型CFTR增效劑CP-628006,這種小分子藥有更高的單個G551D-CFTR激活效率和更持久的F508del-CFTR激活效應,與Ivacaftor聯合使用療效更強。
2.2 Hutchinson-Gilford早老癥
Hutchinson-Gilford 早老癥(Hutchinson-Gilford progeria syndrome,HGPS,MIM:176670)呈常染色體顯性(autosomal dominant,AD)遺傳,患病率為1∶4 000 000,致病基因是LMNA基因[25]。HGPS 患者多于1 歲前發病,面容酷似老人,壽命一般不超過14 歲[25]。LMNA基因編碼核纖層蛋白A、C,纖層蛋白A 可與早老蛋白結合。基因突變產生的早老蛋白一方面在核膜被法尼基化和甲基化產生堆積[26],另一方面使微管與細胞核過度耦聯阻止肌動蛋白移動[27],這些改變共同造成細胞核結構和功能的損害。
2020 年11 月,法尼基轉移酶抑制劑洛那法尼(ZokinvyTM)獲得FDA 治療HGPS 的批準[28],通過阻止早老蛋白法尼基化發揮作用[29]。化合物UCM-13207使早老蛋白從核膜分離以降低其濃度,并有效減少細胞DNA 損傷以恢復細胞功能[30]。結合抑制劑JH4 通過阻止早老蛋白-纖層蛋白A 結合以改善核變性[31]。Remodelin 是N-乙酰轉移酶-10 的新型強效選擇性抑制劑,通過抑制N-乙酰轉移酶-10介導微管重組而補救細胞核形狀的破壞[32],可延長HGPS患兒的壽命[33]。
3 單克隆抗體治療
治療性單克隆抗體(monoclonal antibody,mAb)是一種由B淋巴細胞產生的能高度特異識別抗原的抗體類藥物,包括抗體片段、Fc 融合蛋白及抗體-藥物耦合物,通過遞送細胞毒素、募集細胞和蛋白質及調節信號通路等發揮作用[34]。
自身免疫性多內分泌腺病綜合征I型伴或不伴可逆性干骺端發育不良(autoimmune polyendocrine syndrome, type I, with or without reversible metaphyseal dysplasia, APS1, MIM: 240300)呈AD或AR 遺 傳 , 患 病 率 為 1∶ 1 000 000~9∶1 000 000[35]。APS1 表現為慢性皮膚黏膜念珠菌病、甲狀旁腺功能減退癥和腎上腺皮質功能衰竭三聯征,常常導致患兒的過早死亡[36]。
APS1 的致病基因為AIRE基因,表達于胸腺CD45-MHC-Ⅱ+細胞[35]。AIRE基因突變導致自體反應性T細胞逃避免疫細胞的陰性選擇,引起多系統自身免疫[37]。一項研究發現抗CD45RC mAb 可抑制T 細胞毒效應和恢復Treg 細胞,有效延緩APS1進程,揭示了CD45RC mAb 藥物在APS1 預防性治療中的重要性[38]。
4 基因療法
基因療法以替換、轉入或編輯疾病基因等方式操縱遺傳物質[39],成為近年來孤兒藥的重點研究方向之一[40]。另外,二代測序在臨床遺傳診斷中廣泛應用,CRISPR 技術迅猛發展,也極大地推動了基因治療在孤兒藥研發領域中的應用。CRISPR 是原核生物基因組內的一段重復序列,通過Cas酶特異性切割和修復目標DNA序列,實現目標基因敲除和堿基編輯,是一種更為經濟、迅速且簡單的基因編輯工具[41]。
基因治療按治療途徑的不同可分為體內治療和體外治療2 種[42]。體內治療指通過局部注射攜帶目的基因的病毒載體進行基因編輯,病毒載體多用非整合型病毒[如腺相關病毒(adenoassociated virus,AAV)][43]。體外治療利用自體細胞或同種異體細胞在體外轉入目的基因并進行修飾,也稱為細胞療法[44]。
4.1 基因治療
4.1.1 Leber 先天性黑蒙 Leber 先天性黑蒙(Leber congenital amaurosis,LCA)是基因治療最早成功的案例[37]。LCA 是嚴重的遺傳性視網膜營養不良癥,患病率為1∶81 000~1∶30 000,多于兒童早期發病[45]。LCA 已知至少有28 種基因突變參與致病,遺傳病因占全部病例的75%[46]。RPE65基因是LCA 的重要致病基因,RPE65基因編碼類視黃醇異構酶,負責維生素A在類視黃醇循環中的代謝[45]。
Voretigene neparvovec是AAV2載體相關的基因治療藥物[46],于2017 年被FDA 正式批準用于RPE65雙等位基因突變引起的LCA[47]。AAV2載體可將有編碼類視黃醇異構水解酶功能的RPE65基因導入LCA 患者體內,該藥物通常通過玻璃體切割術注入視網膜間隙[47]。
4.1.2 杜氏肌營養不良 杜氏肌營養不良(Duchenne muscular dystrophy, DMD, MIM:310200)是一種X連鎖肌肉萎縮性疾病,在男性新生兒中的患病率為1∶5 000[43]。DMD 通常于3 歲前發病,出現進行性肌無力和肌疲勞,最終發展為呼吸衰竭和心肌病,常于20歲前死亡[48]。該病由DMD基因突變引起其編碼的抗肌萎縮蛋白(dystrophin,Dys)缺乏[44],Dys 是抗肌萎縮蛋白結合蛋白復合物的關鍵成分,對維持肌纖維剛度至關重要[49]。
Gange 等[47]設計了一種微抗肌萎縮蛋白轉基因(AAVrh74.MHCK7.micro-dystrophin),該藥物以AAV作為載體進行基因轉移,可有效恢復Dys的表達。另一項研究設計出一種經密碼子優化的合成轉基因,編碼微型化的Dys 相關蛋白utrophin(μUtro),μUtro 具有與Dys 相似的結構和功能[50]。該研究對Dys 缺陷的新生mdx 小鼠應用AAVμUtro,發現DMD小鼠模型肌壞死、再生組織和生化標志物的異常在成年之前得到完全控制[51]。
隨著AAV 基因治療向人體臨床試驗的逐步推進,其在轉導效率、組織趨向性及免疫原性等方面的要求也不斷增加,這推動了更為安全、高效的AAV載體的研發。
4.2 細胞治療
Wiskott-Aldrich 綜 合 征 (Wiskott-Aldrich syndrome,WAS,MIM:301000)是一種X 連鎖免疫缺陷病,由WAS基因失功能突變引起,患病率為1∶100 000[52]。WAS 表現為免疫缺陷、血小板減少和濕疹三聯征,常在10歲之前死亡[52]。WAS基因編碼WAS 蛋白,對細胞信號轉導和免疫突觸形成至關重要[53]。
WAS 的一線治療是同種異體造血干細胞移植[53],但常由于供體不匹配使得患兒病死率增加[54],移植物抗宿主病對此類疾病的治療效果有著巨大影響[55-56]。
一項研究首次開發了一種基于CRISPR/Cas9基因編輯平臺的造血干細胞和祖細胞藥物,在患者來源的造血干細胞和祖細胞中敲入治療性WAS cDNA及其內源性翻譯起始密碼子,在60%的患者中得到滿意效果[54]。
4.3 寡核苷酸治療
寡核苷酸治療通過設計特定寡核苷酸序列,以沃森-克里克堿基配對原則為基礎,下調或修飾致病基因、阻斷RNA 翻譯相關蛋白等發揮作用[57]。
寡核苷酸治療可分為2種藥物,包括反義寡核苷酸(antisense oligonucleotide,ASO) 和小干擾RNA。ASO 又可分為核糖核酸酶H1(ribonuclease H1,RNase H1)依賴性ASO 和gapmer[58]。ASO 是一種人工合成的單鏈小分子核酸聚合物,可激活RNase H1裂解目標mRNA以改變mRNA的表達[59]。RNase H1 是一種內源性核酸內切酶,結合同源性mRNA后在ASO結合位點切割靶mRNA調節基因表達[58]。小干擾RNA 的作用機制是引導沉默復合物中的Argonaute 2 蛋白結合目標轉錄物,引起基因沉默[58]。
4.3.1 DMD 2016年9月,Eteplirsen(Exondys51)獲得FDA 的加速批準,是第一種獲批治療DMD 的磷酸二酰胺嗎啡寡聚物,其機制與51 號外顯子的跳躍有關[60]。Eteplirsen 可操縱DMD 的Dys 轉錄本剪接并使閱讀框得到恢復[61]。一項觀察性研究證明了Eteplirsen 對無法行走或在治療過程中失去行走能力的患兒的潛在益處[60]。
Golodirsen(SRP-4053)是一種新型磷酸二酰胺嗎啡寡聚物,于2019 年被FDA 批準,與Dys 的前體mRNA 互補并恢復該mRNA 的閱讀框,與53號外顯子的跳躍有關,其安全性、藥代動力學和生物活性均得到了驗證[62]。
4.3.2 Batten 病 Batten 病也稱神經元蠟樣質脂褐質沉積癥,是一組包含13 種神經退行性疾病的遺傳性疾病,是兒童癡呆癥最常見的形式[63]。
對于該疾病的特定基因突變,研究者為一位神經元蠟樣質脂褐質沉積癥7型兒童制定了為期一年的“N-of-1”個性化治療方案[64]。研究團隊設計出了一種ASO藥物——Milasen,該藥物靶向患兒6號外顯子剪接到6號內含子的隱秘剪接位點及其附近剪接增強劑[64]。經過Milasen治療,患兒癲癇發作頻率和持續時間都下降至少50%。該藥物的使用為孤兒藥的個性化治療提供了可參考依據,也為個性化基因組醫學提供了范例。
5 “超適應證”使用治療藥物和老藥新用
“老藥新用”指發現了某已知藥品的新性質或功能并用于新領域。再利用藥物的安全性和質量在先前研究和長期使用中均已得到驗證,其不良反應發生的風險相對較低,可更快開始臨床試驗,對于罕見病的治療更加有利。
5.1 普萘洛爾治療Lafora肌陣攣性癲癇
Lafora 肌陣攣性癲癇(Lafora disease,LD,MIM:254780)是一種致命的神經退行性疾病,患病率小于1∶1 000 000[65],特征是患兒全身癲癇發作和快速進展為植物狀態。LD由EPM2A或EPM2B基因突變引起[66]。
普萘洛爾最初用于高血壓、特發性震顫和焦慮。證據表明,普萘洛爾抗血管生成、促凋亡和抗炎特性也可應用在不同的罕見病中[67-68]。一項動物實驗研究表明,普萘洛爾作為炎癥調節劑,可減少星形膠質細胞和小膠質細胞炎癥化,對LD早期治療有潛在效果[66]。
5.2 除草劑治療尿黑酸尿癥
尿黑酸尿癥(alkaptonuria, AKU, MIM:203500)呈AR遺傳,患病率為1∶250 000[69],由編碼尿黑酸1,2雙加氧酶的HGD基因突變引起,嬰兒期即可起病[70]。該病主要為酪氨酸代謝障礙引起,酪氨酸參與兒茶酚胺神經遞質的生物合成,對神經的興奮和抑制起重要作用[71]。
尼替西農最初被作為除草劑開發,后來被發現可以用于抑制尿黑酸堆積并延緩AKU 的疾病進展。一項研究報告了一組AKU 患者在尼替西農治療試驗前后24 h 的變化,發現用藥后患者尿液中神經代謝物去甲腎上腺素減少,腎上腺素、3-甲氧酪胺增多,證明尼替西農可使AKU 患者的神經遞質代謝得到改善[69]。
6 小結
綜上,在過去的十年中,生物制藥行業在為罕見病群體提供新療法方面取得了一定進展。但孤兒藥的開發仍然受有限的罕見病人數限制,也受高昂的研發和治療費用掣肘。為加快開發罕見病新療法的進程,未來需要投入更多關注和出臺更好的激勵措施。
利益沖突聲明:所有作者均聲明不存在利益沖突。
猜你喜歡
英語世界(2023年6期)2023-06-30 06:29:10
健康體檢與管理(2022年2期)2022-04-15 22:33:17
昆明醫科大學學報(2021年1期)2021-02-07 01:06:44
中國生殖健康(2020年2期)2021-01-18 02:51:26
小學生導刊(2018年13期)2018-06-29 03:49:00
現代檢驗醫學雜志(2016年4期)2016-11-15 02:01:14
中國男科學雜志(2016年9期)2016-03-20 15:00:09
中國健康心理學雜志(2015年5期)2015-09-05 09:55:52
中國當代醫藥(2015年22期)2015-03-01 02:05:33
中國神經精神疾病雜志(2014年1期)2014-03-01 03:23:22
