不同禁帶寬度催化劑協同低溫等離子體氧化NOx
2023-09-14 05:14:12郭子妮屈吉艷羅建洪
無機鹽工業 2023年9期
關鍵詞:催化劑
郭子妮,屈吉艷,羅建洪
(四川大學化學工程學院,四川成都 610065)
隨著社會發展,氮氧化物的排放導致臭氧層的破壞、引起光化學煙霧和酸雨、加劇溫室效應等環境問題[1],控制氮氧化物的排放屬于當務之急。國內外采用改進燃燒技術和用特定的物理化學方法控制NOx排放[2-3]。其中氧化吸收法中的氣相催化氧化法已非常成熟,催化氧化法包括金屬氧化物催化氧化[4]、光催化氧化[5]和Fenton體系催化氧化[6]等。
金屬氧化物是在NO 催化氧化領域研究較早、發展較快的一種催化劑,傳統金屬氧化物催化氧化法是以煙氣中多余的氧氣作為氧化劑,以負載在活性炭、Al2O3和SiO2上的V、W、Ti 及稀土金屬氧化物為催化劑。金屬氧化物催化氧化法已廣泛應用于煙氣凈化工藝,脫氮效率可達90%以上[4]。WANG等[7]通過水熱法合成了不同價態的錳氧化物,在48 000 mL/(g·h)的空速條件下,以氧氣為氧化劑時,NO 的最大轉化率可以達到91.4%。MA 等[8]通過可調控水熱法合成了納米顆粒狀SmMn2O5單晶;其中,類納米顆粒SmMn2O5在300 ℃條件下能實現90%以上的NO 轉化率。等離子體技術設備簡單、操作便捷。LIU 等[9]通過非熱等離子體技術制備了Mn-O-Ce催化劑,并且在催化劑中形成了Mn-O-Ce缺陷相,發現非熱等離子體處理后的催化劑表現出活性顯著增強、比表面積增加、孔體積增大,此外還改變了催化劑表面活性粒子的相對表面濃度及其氧化態。ZHANG 等[10]在合成過程中用等離子體法處理得到吸附性質較好的NiO-TiO2-Al2O3材料,處理后的催化劑比表面積增加,樣品中的Ni顆粒的分散性也明顯改善,所以復合物具備較強的NOx吸附能力,從而促使NOx的處理效率進一步得以提高。等離子體協同催化是等離子體和催化劑結合之后在反應系統中發揮協同作用。LI 等[11]研究了等離子體活化與4 種銅基催化劑混合體系對NO 和CH4的脫除,闡明了12%CuO/10%CeO2/15%TiO2/γ-Al2O3催化劑具有較低的NO解吸溫度、較大的峰面積,導致其對NO+CH4脫除反應的催化活性比其他催化劑更好。通過使催化劑與低溫等離子體協同,反應全程溫度變化不大且接近室溫,更換不同的催化劑可同時凈化多種污染物。
但是在常見催化劑協同低溫等離子體的相關報道中,不同催化劑在此過程中產生影響的對比分析,并沒有做過多深入的闡述。本研究采用溶劑熱法、煅燒法、沉積法等制備了WO3、ZnO、NiO、CdS 和Cu2O 共5 種禁帶寬度不同的半導體催化劑,對比了催化劑協同低溫等離子體催化氧化NO 與NOx的性能,說明各自對反應過程的具體影響,以及等離子體與各催化劑之間的耦合作用。并且本文結合已非常成熟的堿液吸收形成閉環,降低二次污染,將催化后的尾氣再通過堿液吸收達到國家標準,結晶得到的硝酸鹽比直接還原有更高的商業價值。經過放電催化后的樣品,借助離子色譜測定其表面的NO3-與NO2-的含量以及穩定性測試等,有利于進一步闡釋二者的協同機制。
1 實驗部分
1.1 試劑和儀器
試劑:二氧化鈦、鎢酸銨、無水乙醇、硝酸鉀、十六烷基三甲基溴化銨(CTAB)、氧化鋁、N,N-二甲基甲酰胺、硅溶膠、一氧化氮、氨水、六水合硝酸鋅、尿素、硫脲、硫化鎘、乙酸鎳、三乙醇胺、乙二醇、醋酸銅,均為分析純;高純氮氣、高純氧氣。
儀器:LM-50-200ML型均相反應器;DZF-1ASB型真空干燥箱;CTP-2000K 型等離子體發生電源;KM9206 型便攜式煙氣分析儀;TS-100B 型恒溫振蕩器。
1.2 不同禁帶寬度催化劑的制備
WO3的制備:采用水熱法和高溫煅燒法相結合制備WO3。稱取8.528 0 g CTAB于70 mL蒸餾水中,在磁力攪拌下將1.921 4 g 鎢酸銨溶解于混合溶液中,在室溫下用氨水將其pH 調至8.5 左右。轉移到150 mL 高壓反應釜中,于140 ℃下反應24 h。得到白色固相,用去離子水清洗3次,在105 ℃干燥12 h,得到白色塊狀樣品。將樣品研磨為粉末狀放入600 ℃高溫節能箱式爐中煅燒10 h,升溫速率為5 ℃/min。
ZnO 的制備:采用溶劑熱法合成 ZnO 催化劑。稱取六水合硝酸鋅7.600 0 g,溶解于150 mL 乙醇-水溶液,其中無水乙醇與去離子水的體積比為1∶2,磁力攪拌20 min 至溶液呈無色透明,向溶液中加入3.500 0 g尿素后繼續攪拌1 h,轉入250 mL聚四氟乙烯反應釜中,在150 ℃下反應16 h。自然冷卻,經無水乙醇和去離子水的多次洗滌,高速離心分離后獲得白色固體沉淀物,在105 ℃干燥12 h,最后將其放入550 ℃高溫箱式爐中煅燒5 h。
CdS 的制備:采用溶劑熱法合成 CdS 催化劑。將8 mmol 硫脲和2 mmol CdCl2分散在60 mL 由乙醇和N,N-二甲基甲酰胺(體積比為3∶1)組成的混合溶液中,磁力攪拌1 h。將前驅物溶液轉移到100 mL內襯特氟隆的高壓釜中,在 150 ℃下加熱反應12 h。自然冷卻,用大量的去離子水和無水乙醇多次洗滌,于60 ℃下真空干燥24 h。
NiO 的制備:采用均勻沉淀法制備NiO。將2.366 5 g四水合乙酸鎳和2.411 7 g尿素分別溶解于200 mL 去離子水中,將兩種溶液混合在一起,向其中逐滴加入氨水以調節pH 至10 左右,隨后在80 ℃下保持6 h,經過3 次清洗后干燥,放在箱式爐中400 ℃下煅燒3 h,得到黑色NiO樣品。
Cu2O 的制備:采用溶劑熱法合成Cu2O。量取15 mL 三乙醇胺逐滴加入到45 mL 乙二醇中,加入1.800 0 g 醋酸銅,磁力攪拌1 h,將混合液轉移到150 mL 高壓反應釜中于160 ℃下反應1 h。自然冷卻,用無水乙醇和去離子水洗滌3遍,離心分離出沉淀物,將其放入80 ℃干燥箱內烘干12 h,得到Cu2O樣品。
1.3 催化劑與載體的混合
稱取1.500 0 g 各催化劑樣品,將其分別與15.000 0 g 近似球形顆粒(直徑約為2.0 mm)的γ-Al2O3混合,在25 ℃下以350 r/min 的轉速振蕩15 min,使催化劑緊密附著在γ-Al2O3顆粒表面上。選用不銹鋼篩子對其依次篩選,得到附著均勻且牢 固 的WO3/γ-Al2O3、ZnO/γ-Al2O3、CdS/γ-Al2O3、NiO/γ-Al2O3和Cu2O/γ-Al2O3混合物待測試。
1.4 實驗流程
實驗室自行搭建的低溫等離子體氧化NOx的實驗裝置如圖1所示。
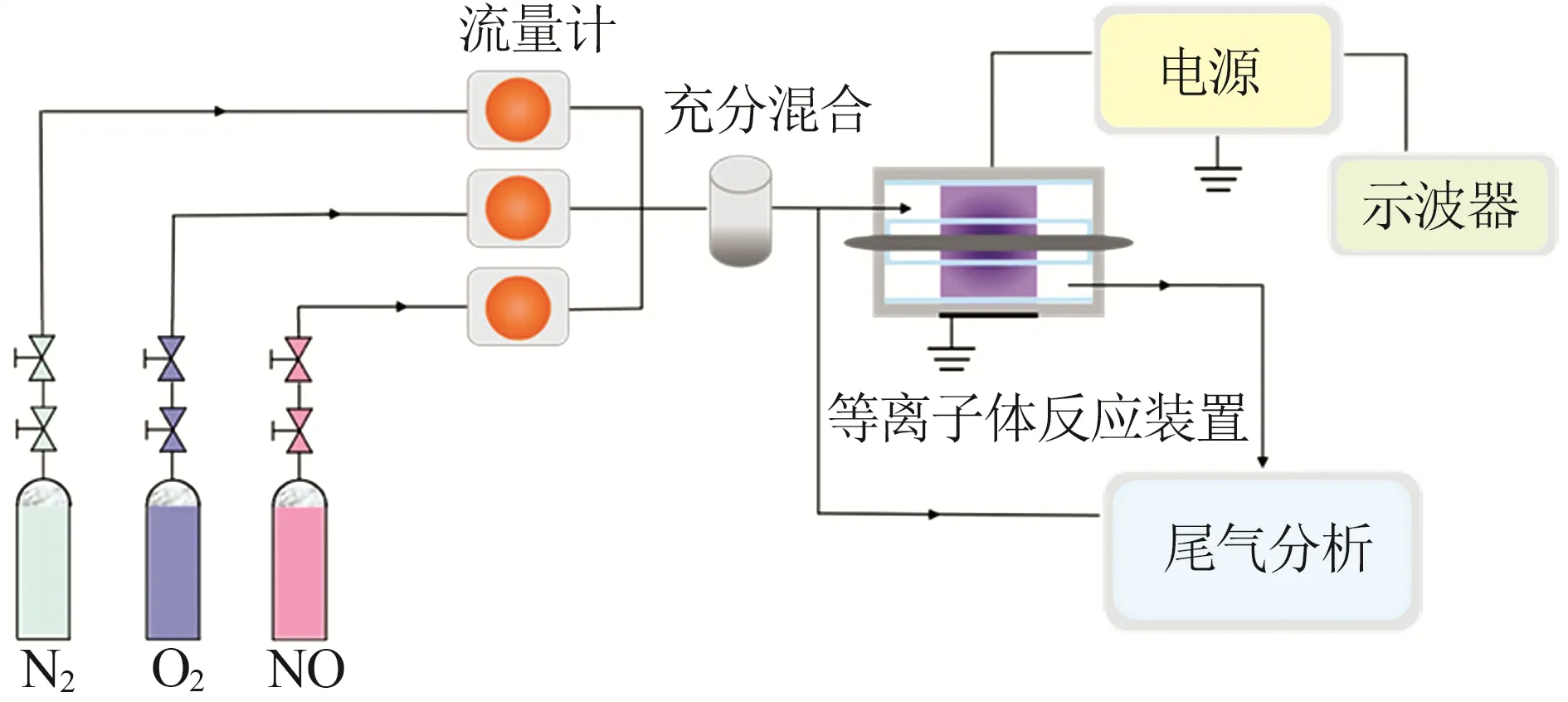
圖1 等離子體脫硝的實驗裝置示意圖Fig.1 Schematic diagram of experimental setup for plasma(co-catalyzed) denitration
1)進氣部分:充分混合后的N2、O2和NO通入雙介質阻擋等離子體反應器內。設定氣體總流速為1.5 L/min 左右,混合氣中的NO 體積分數為(500±10)×10-6、O2為6%±0.1%(體積分數)。
2)電源部分:反應體系中由CTP-2000K 型低溫等離子體發生電源進行調控,采用0~30 kV 的脈沖調制電源,其調制脈沖頻率范圍在1~1 000 Hz;脈沖占空比為1%~99%。本研究中,設定脈沖頻率為75 Hz,固定脈沖占空比為70%。放電反應產生的電壓和電流由TBS-1000B 型數字示波器通過高壓探頭和電流探頭測定記錄放電時的波形圖像。
3)等離子體協同催化氧化NOx部分:等離子體放電反應器內的放電過程由脈沖調制電源控制。采用雙介質同軸DBD 反應器,通過不同種類的催化劑與低溫等離子體的協同作用使得NOx高效氧化。附著后的催化劑均勻填充在放電區間的石英管中。
4)尾氣分析部分:采用KM 9206 型便攜式煙氣分析儀檢測尾氣的NO含量、NOx含量以及氧含量。
2 結果與討論
2.1 材料表征
2.1.1 XRD分析
采用X 射線衍射分別對WO3、ZnO、NiO、CdS 和Cu2O進行分析,如圖2所示。圖2a中根據JCPDS 數據庫的標準卡片(JCPDS 43-1035)得知,WO3粉末樣品在23.1°、23.6°和24.3°處出現特征峰分別對應晶面(002)(020)(200),這說明通過此煅燒法制備獲得的是單斜相WO3。由圖2b 可知,ZnO 粉末樣品依次在31.7°、34.4°、36.2°、47.5°、56.5°、62.8°、67.9°、69.0°處出現明顯特征峰分別對應ZnO 的(100)(002)(101)(102)(110)(103)(200)(112)晶面,發 現這與ZnO 的XRD 標準卡片(PDF 36-1451)一致,證實了所制得樣品是六方纖礦型ZnO。由圖2c 的NiO 的XRD譜圖可以知,處于37.3°、43.3°、62.9°、75.4°位置處的峰,分別與標準卡片(JCPDS File No.89-5881)上出現的(222)(400)(440)(622)這4 個晶面對應,與NiO 的立方結構實現吻合。由圖2d 可以看到在2θ為 24.8°、26.5°、28.2°、36.7°處有與(100)(002)(101)(102)晶面對應的衍射峰,這與JCPDS No:41-1049 標準峰的位置對應,證明合成的CdS 樣品屬于CdS 六方纖鋅礦相。催化劑Cu2O 的XRD 譜圖見圖2e,明顯得知在29.6°、36.4°、42.3°、61.4°、73.6°處都有相對弱一些的衍射峰,分別對應于晶面(110)(111)(200)(220)(311),并沒有發現有單質銅的對應峰出現,這樣所得到的Cu2O 樣品純度相對較高。
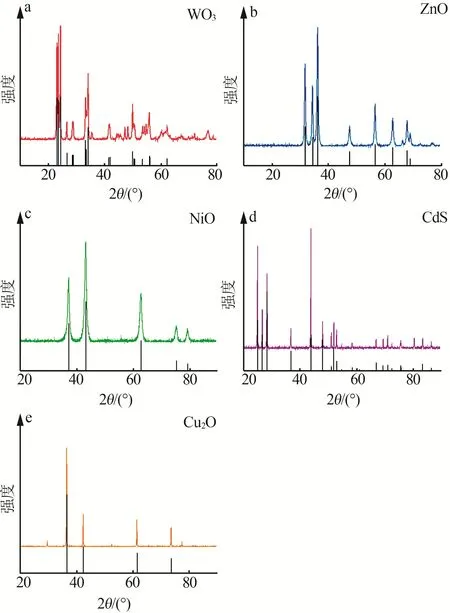
圖2 WO3(a)、ZnO(b)、NiO(c)、CdS(d)和Cu2O(e)的XRD譜圖Fig.2 XRD patterns of WO3(a),ZnO(b),NiO(c),CdS(d) and Cu2O(e)
2.1.2 XPS分析
圖3 為WO3、ZnO、CdS、NiO 和Cu2O 的XPS 譜圖。由圖3a 可知,結合能為35.85 eV 和38.00 eV 的WO3雙峰分別對應于W 4f7/2和W 4f5/2態。這可能歸因于來自+6價氧化態的W原子的光電子,在WO3氧化態附近存在氧空位和表面缺陷。WO3的O 1s光譜在530.70、530.90、532.00 eV 處具有單個不對稱峰,如圖3b 所示。530.70 eV 的峰對應于WO3中的氧原子,530.90 eV 的峰歸因于WO3晶體結構中的缺陷氧O2-,532.00 eV 的峰與吸附在樣品表面上的氧或者水分子有關[12],清楚地表明樣品中都存在缺陷和氧空位。圖3c顯示了Zn特征峰位于1 022.35 eV和1 045.55 eV,分別與Zn 2p3/2和Zn 2p1/2對應。此外,圖3d 顯示了在531.15 eV 的O 1s 對應峰,這是由于催化劑ZnO內的缺氧區域中存在松散結合的氧。圖3e顯示了 Cd 3d5/2和Cd 3d3/2在402.70 eV和409.45 eV處的兩個特征峰。以159.15 eV和160.25 eV為中心的峰擬合到圖3f中,分別是S 2p3/2和S 2p1/2。圖3g描繪了Ni 2p在870.4 eV和851.5 eV處表現出的雙峰,分別對應于Ni 2p3/2和Ni 2p1/2,這是由于自旋軌道耦合效應而形成[13]。由圖3i 可知,在931.1 eV 和950.0 eV處顯示兩個主要峰,可以歸因于Cu+的分峰Cu 2p3/2和Cu 2p1/2[14]。圖2e的XRD譜圖充分證明了Cu2O 的純度較高,圖3i 的XPS 譜圖證明了金屬Cu存在價態為+1價。
2.1.3 UV-Vis分析
圖4顯示了5種不同樣品的UV-Vis譜圖。從圖4a 中觀察到樣品的光吸收起始位置都不同,WO3、ZnO、NiO、CdS 和Cu2O 分別約為450、390、350、550、450 nm,其中ZnO 的吸光度表現最強,Cu2O 最弱。由圖4b 可知,5 種樣品的禁帶寬度分別約是2.81、3.34、3.63、2.40、2.21 eV,這個光學帶隙值與多篇報道內容[15-16]是相對一致的。
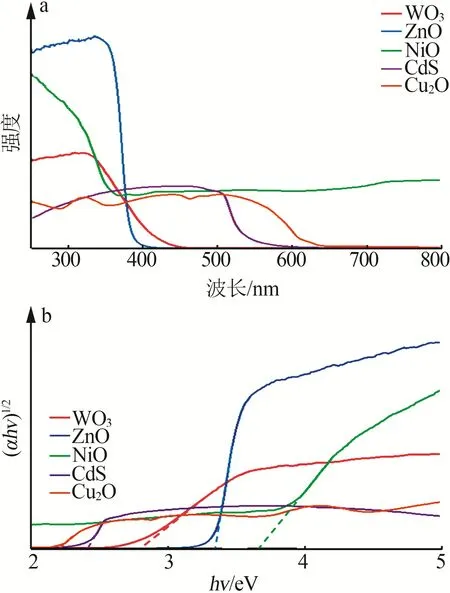
圖4 WO3、ZnO、NiO、CdS、Cu2O紫外可見光譜Fig.4 UV-Vis spectra of WO3,ZnO,NiO,CdS,Cu2O
2.2 催化劑與等離子體之間的相互作用
2.2.1 催化劑對等離子體放電的影響
空管放電過程中,大量均勻分布的亮絲線意味著在這個輸入功率下,等離子體系統的放電強度充分。CdS/γ-Al2O3填充在反應器后,放電亮度下降,是由于放電間隙中存在介質,縮短了放電間隙,細絲線放電模式被改變為粒子表面放電模式和間隙穿線放電模式的組合[17]。WO3/γ-Al2O3、ZnO/γ-Al2O3、CdS/γ-Al2O3的放電現象沒有明顯區別,放電亮度較低的原因仍然是放電模式的轉變。而NiO/γ-Al2O3、Cu2O/γ-Al2O3填充在反應器中未被激活,沒有放電現象,因而沒有顯現出催化性。
2.2.2 等離子體放電對催化劑的影響
為了探究等離子體放電后的各半導體催化劑晶相是否穩定,對反應后的WO3、ZnO和CdS的晶型進行了表征,發現與反應前無差別,說明在等離子體場中制備的半導體的晶型結構是穩定的,經過放電后仍保持較高的結晶度。催化劑的形貌顏色是初步判斷其光響應特性的依據,對反應后催化劑的表觀進行了比對,發現WO3、ZnO和CdS三者的顏色在反應前后均沒有明顯變化。
2.3 不同禁帶寬度催化劑協同低溫等離子體氧化NOx性能的考察
2.3.1 電源電壓對協同催化氧化NOx的影響
等離子體反應過程中,衡量反應器輸出能量的必要指標之一就是能量密度(SIE)。圖5 闡明了DBD反應器中(脈沖)電源電壓的峰值與SIE之間的相互影響,電壓與SIE 的相互關聯是正相關。因為電源電壓的增大,放電強度增強,氣體在受到外加的強電場時其電離、激活的可能性及活化程度也均會提高。體系最主要的能量是來自于質量較輕的、被激活后的帶電粒子自身所帶有的能量,SIE 的大小自然會增大。
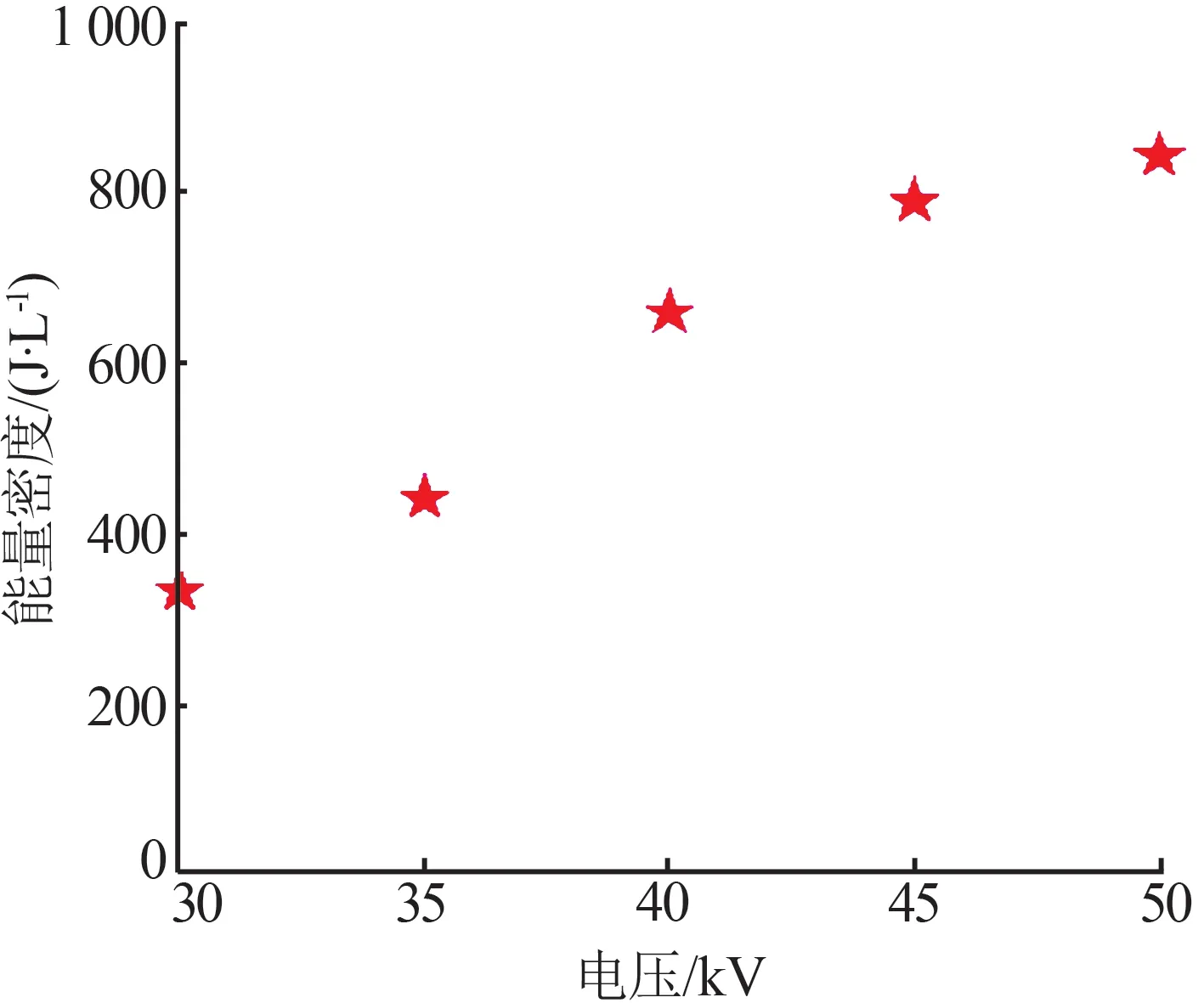
圖5 脈沖調制電源放電中的峰值電壓對能量密度的影響Fig.5 Effect of peak voltage on SIE in pulse-modulated power supply discharge
圖6a、b 是催化劑協同等離子體對NO 與NOx脫除效率在不同電源電壓下的變化。首先,通過對比圖6a、b可知,γ-Al2O3對NO與NOx的氧化效果最差,在電源電壓為40 kV、能量密度為660.46 J/L 時,γ-Al2O3對二者的氧化率為55.7%和36.7%。這是由于γ-Al2O3對氮氧化物的吸附和γ-Al2O3較高的介電常數導致的電場強度增加所致。
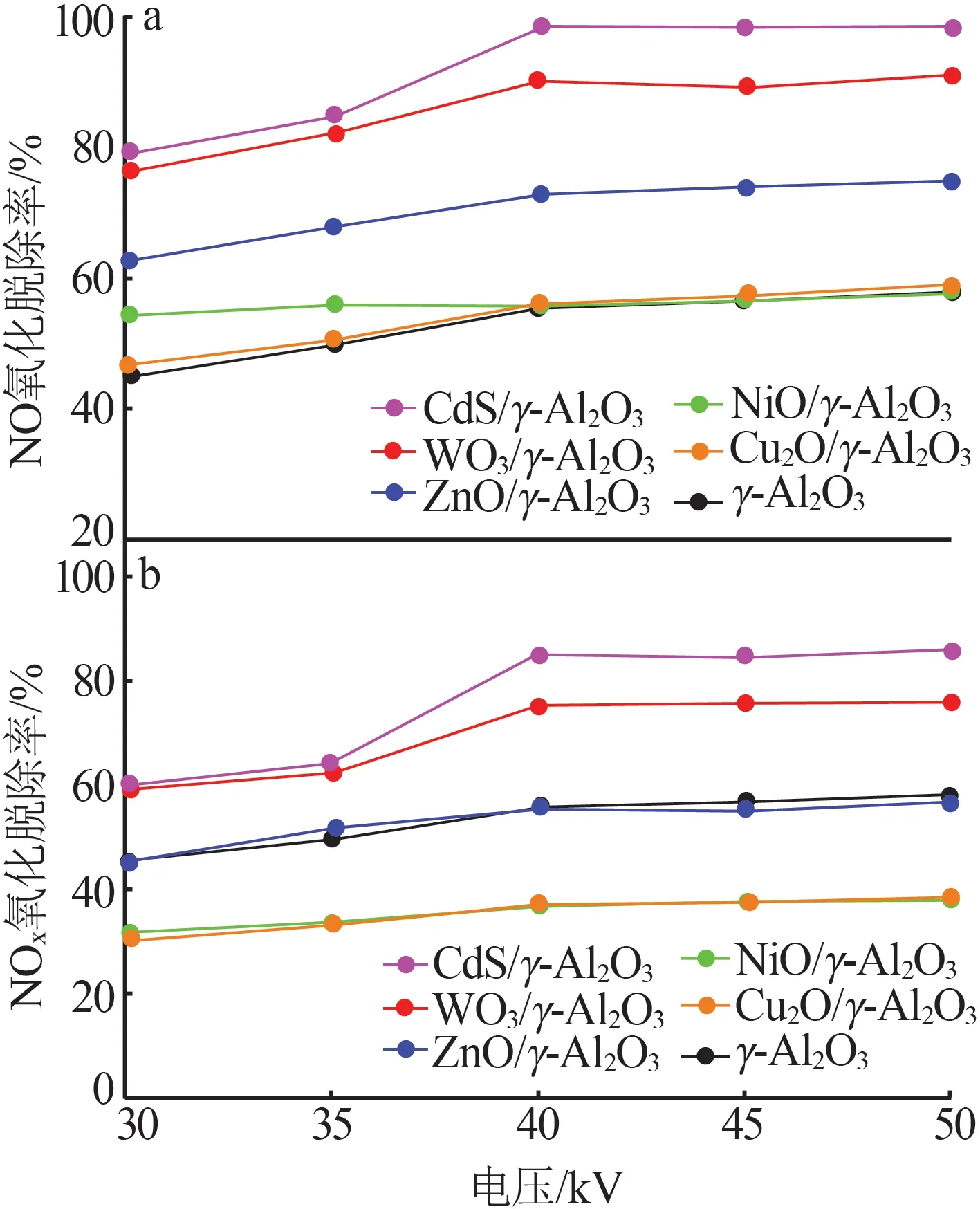
圖6 不同電源電壓對催化劑協同等離子體對NO(a)、NOx(b)氧化脫除率的影響Fig.6 Effect of different power supply voltages on NO(a),NOx(b)oxidation removal efficiency of catalysts in synergistic plasma
在同一電壓下,各催化劑的氧化脫硝效果由強到弱的順序依次為CdS/γ-Al2O3、WO3/γ-Al2O3、ZnO/γ-Al2O3、γ-Al2O3、NiO/γ-Al2O3與Cu2O/γ-Al2O3,后兩者相差不大。CdS催化氧化效率更高可能是較低的禁帶寬使其對激發更敏感,吸收更廣泛的紫外線和可見光波長,為NOx的氧化創造更多“電子-空穴對”。對N型半導體(自由電子濃度遠大于空穴濃度的雜質半導體)來講,同一條件下其氧化脫硝效率與其自身的帶隙能呈負相關,是因為帶隙能小,激活需要的能量少,被活化的幾率就更大,氧化脫硝性能較好。
當輸入SIE 由660.46 J/L 提高至843.62 J/L(即電壓從40 kV 上升到50 kV)時,WO3、ZnO 和CdS 去除NO和NOx的性能沒有得到較大提升,首先是反應器中的氣體處于高能量密度氛圍中,獲取了更高的電子能量,引發了更復雜的N2與O2的反應,其次是在更高的放電功率下,反應區域自然會出現熱量釋放出來的情形,溫度大致為80~100 ℃,這就導致了催化劑從球狀載體γ-Al2O3的表面附著不牢固而引起滑落。
但 對 于P 型 半 導 體,NiO/γ-Al2O3和Cu2O/γ-Al2O3的氧化脫硝能力與單一γ-Al2O3幾乎無差別,NO脫除率依次為56.1%、56.5%;NOx去除率分別為36.9%、37.3%。在同一條件下,其氧化脫硝效率的高低與其帶隙能沒有關聯,在此環境完全得不到激活。P 型半導體(以帶正電的空穴導電為主的半導體)中自由移動、帶電荷的是空穴居多一些,而N型半導體中大多是電子。在等離子體的電場中,這樣的附加能量可以到達電子上而后碰撞轉移能量,進一步促進化學反應進行,所以能夠斷定是電子在此環境下更容易被激活。
綜上所述,脈沖電源提供的電壓越高,獲得的氧化脫硝性能越好,但當電壓上升到一定值后,氧化NO以及NOx效果的提升幅度變慢。圖7為脈沖調制電源放電中的峰值電壓對能量效率η的影響。從圖7也可看出電壓上升到40 kV 之后,整個等離子體放電催化系統的能量效率η出現下降的趨勢,這是因為電源所給予的能量足夠,同時體系中電子也帶有較大的能量,但是反應過程中有大部分能量沒有被利用而以熱能的形式釋放,能量利用效率降低。
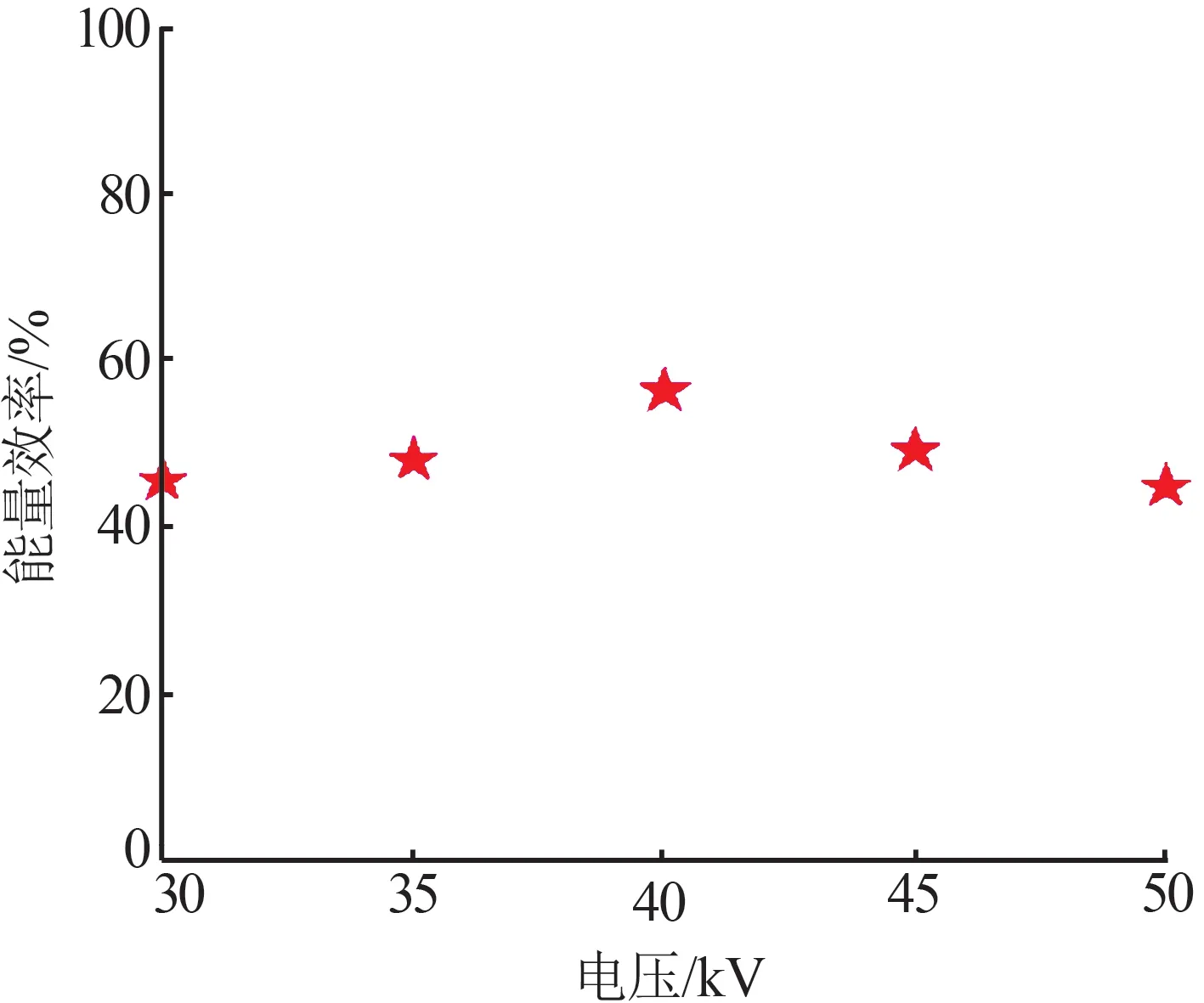
圖7 脈沖調制電源放電中的峰值電壓對能量效率η的影響Fig.7 Effect of peak voltage on energy efficiency in pulse-modulated power supply discharge
2.3.2 離子色譜分析
在低溫等離子體協同半導體催化劑對NOx氧化過程中,氣體將會被氧化為更高價態的物質累積在催化劑表面,長時間反應后催化劑的氧化效果會有下降趨勢,這也是NOx氧化效率呈現出遞減的首要原因。采用離子色譜分析(IC)測定累積在催化劑表層的產物。圖8 為催化劑在40 kV 電壓下放電后表面附著的NO3-與NO2-的量。如圖8 所示,在電源電壓為40 kV 下,不同種類催化劑表面的NO2-與NO3-附著量由大到小的順序為CdS/γ-Al2O3、WO3/γ-Al2O3、ZnO/γ-Al2O3、γ-Al2O3、NiO/γ-Al2O3與Cu2O/γ-Al2O3,后兩者接近,其協同等離子體共同對NOx的氧化效果與上述順序一致,因此,等離子體催化氧化脫硝效率越高,累積在相應催化劑表面的NO2-與NO3-的量就越高。
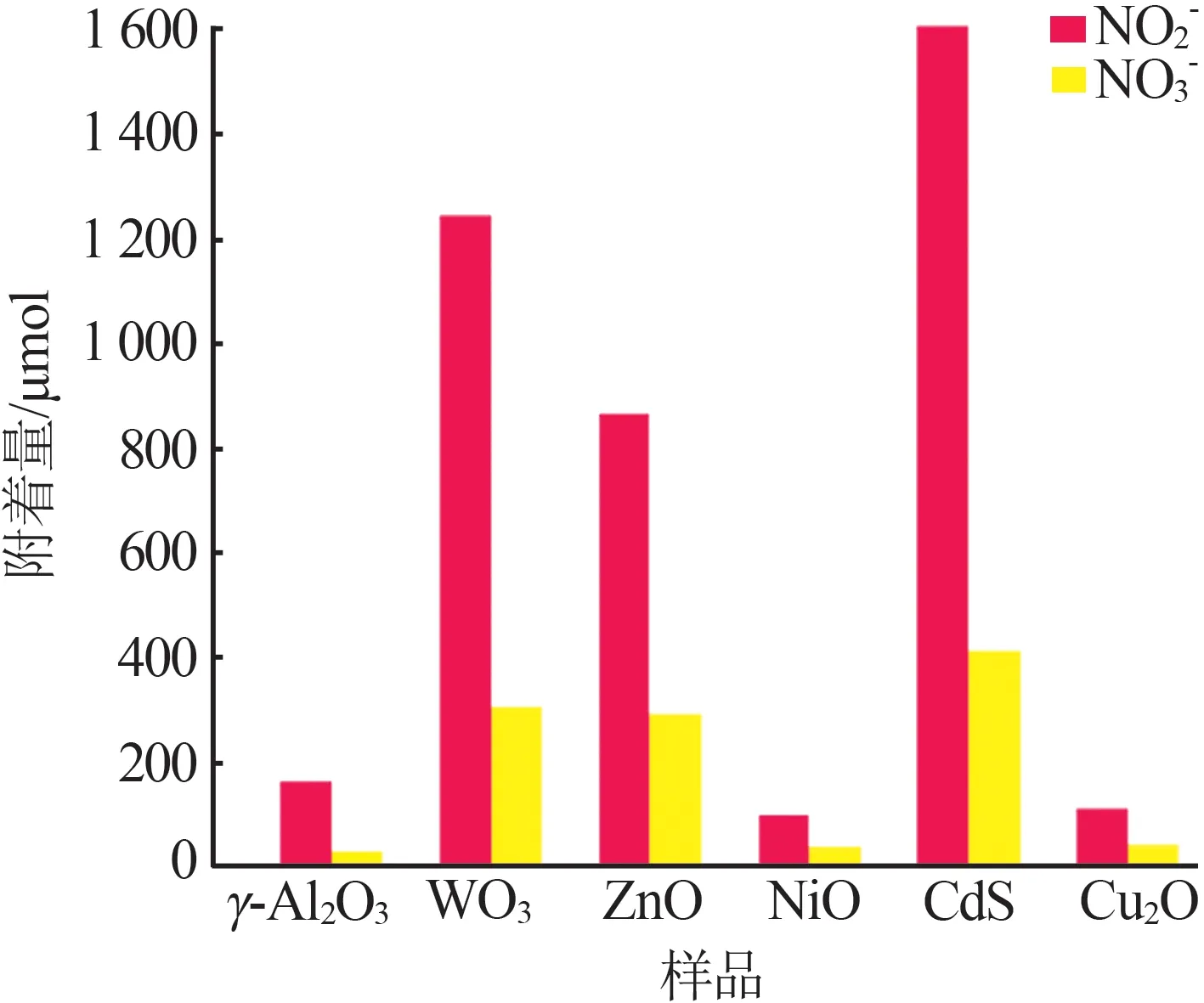
圖8 催化劑在40 kV電壓下放電后表面附著的NO3-與NO2-的量Fig.8 Dosage of NO3- and NO2- attached to surface of catalysts after discharge at 40 kV
單獨的γ-Al2O3填入放電區間,反應后表面的NO2-與NO3-較少,說明該載體催化效果不好。將NiO 和Cu2O 兩類P 型催化劑樣品附著于γ-Al2O3顆粒,反應后表面的NO3-量極低,同時檢測出其表面NO2-的量與未附著催化劑的γ-Al2O3顆粒表面NO2-的量相當,論證了催化劑NiO和Cu2O的催化效果較差。相反,3 種N 型半導體WO3、ZnO 和 CdS 附著在γ-Al2O3顆粒外層,通過放電氧化NOx后其表面的NO2-和NO3-比僅填充γ-Al2O3時的量高很多倍。同時證實了CdS、ZnO 和WO3等N 型半導體催化劑在等離子體場中被激發,實現了NOx的深度氧化。此外,NO3-與NO2-在3 種N 型催化劑表面累積量由大到小依次為CdS、WO3、ZnO,表明CdS比WO3和ZnO對等離子體發生器場的激發更敏感,這是由于CdS 的禁帶寬較窄。由于等離子體放電時產生的紫外光強度基本上不足以激發傳統的光催化過程,因此認為,等離子體協同半導體催化的關鍵因素之一是催化劑是否對等離子體場產生的高能電子的激發更加敏感,而這種現象的產生是因為CdS 的帶隙能比WO3和ZnO低。
2.3.3 等離子體協同半導體催化氧化NOx的穩定性測試
圖9 為協同雙介質阻擋放電氧化NOx的性能測試。由圖9 可知,協同CdS 的雙介質阻擋放電實驗(SIE=660.46 J/L)中,長時間氧化氮氧化物的效果穩定性良好,說明CdS 在長期放電過程中氧化脫硝性能保持良好。圖10 為低溫等離子體協同CdS 催化氧化NOx的性能分析。從圖10a可以看出,在120 min的連續實驗中60 min 時CdS 的平均NO 和NOx氧化率分別為96.8%和82.2%;由圖10b 可知,經過兩次洗滌再生,60 min時CdS分別氧化了93.5%和78.2%的NO 和NOx,與同一時間時的氧化率對比,出現下降趨勢可能的原因是納米CdS受到了放電時伴隨的紫外可見光的腐蝕影響,這為DBD 驅動的CdS催化氧化NOx的工業應用提供了良好基礎,有較好的經濟效益。圖10中空缺處為分析設備自清潔。
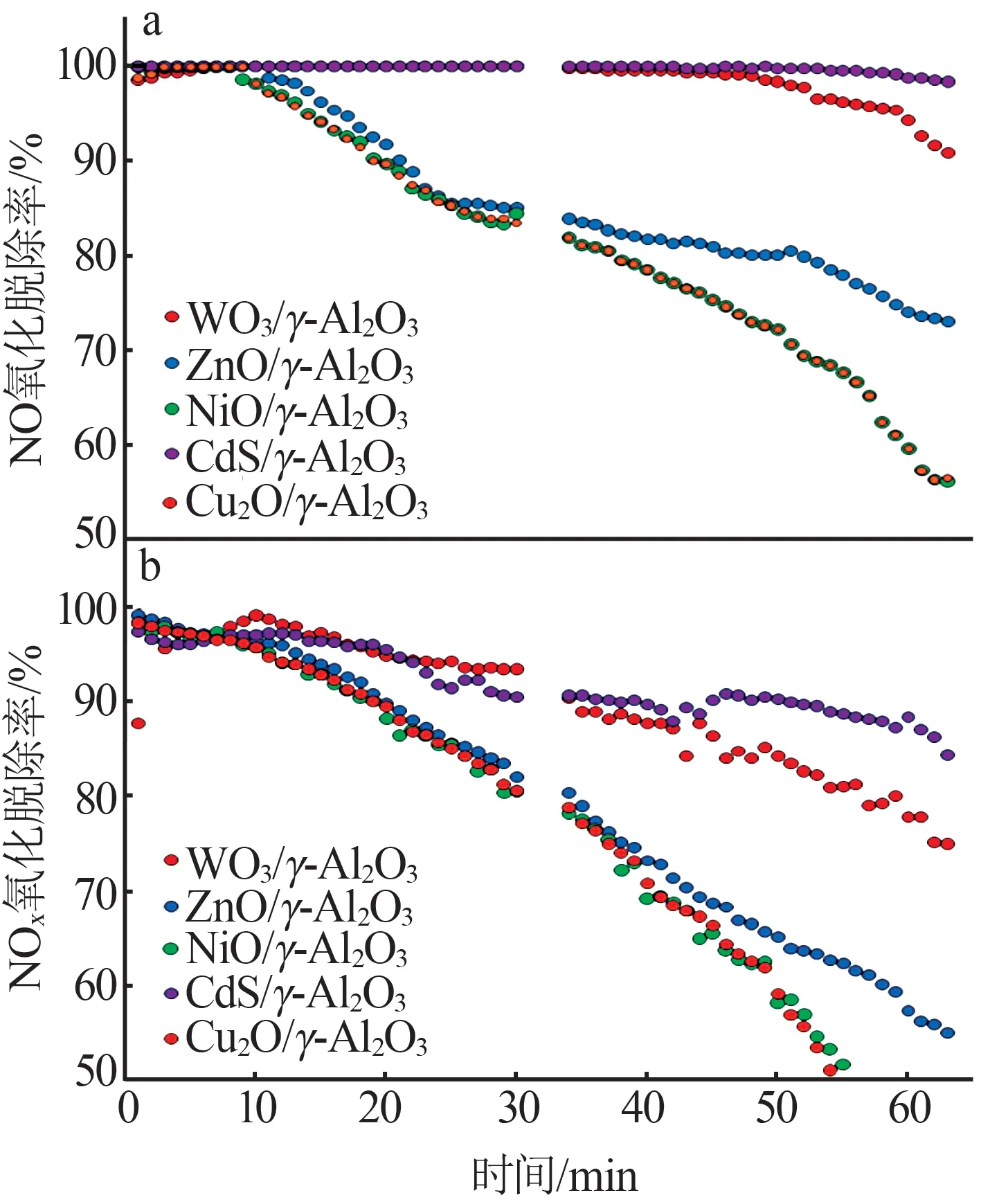
圖9 γ-Al2O3、WO3、ZnO、NiO、CdS和Cu2O協同雙介質阻擋放電氧化NO、NOx的性能測試Fig.9 Performance test of long-term NO,NOx oxidation by γ-Al2O3,WO3,ZnO,NiO,CdS and Cu2O with dual dielectric barrier discharge
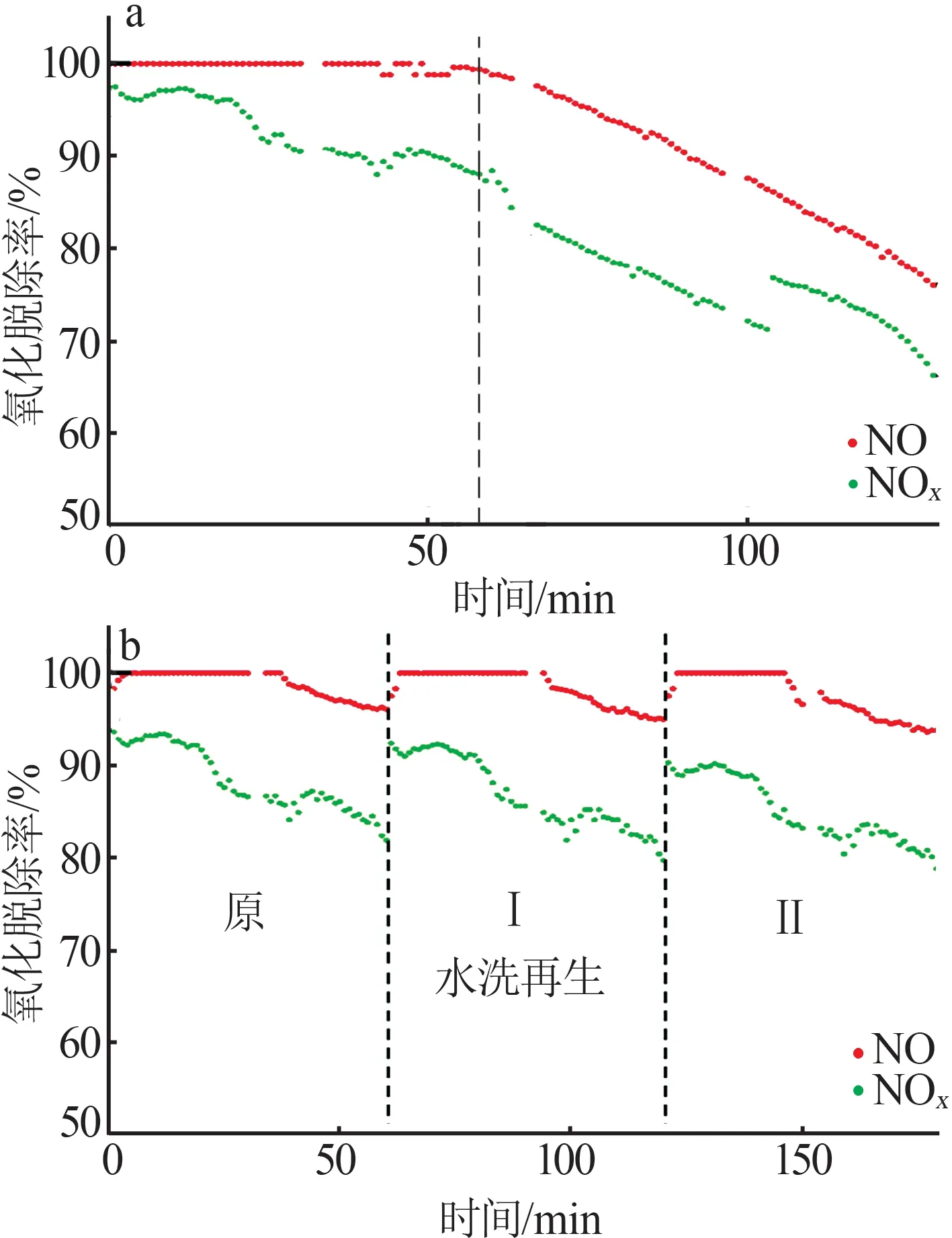
圖10 低溫等離子體協同CdS催化氧化NOx、NO性能分析Fig.10 Performance analysis of low temperature plasma synergistic CdS catalytic oxidation of NOxand NO
3 反應機理
等離子體協同CdS催化氧化NOx可能的反應機理如圖11所示。首先,等離子體提供大量的高能電子激發CdS產生電子-空穴對(e--h+),電子與空穴分別與吸附在CdS表面的H2O與O2反應生成大量·OH和·O2-。其次,吸附在CdS 表面的NO 被產生的·OH氧化成NO2,然后進一步深度氧化成NO2-與NO3-,吸附在CdS 表面的NO2則直接被產生的·OH 氧化為NO2-與NO3-。最后,在放電激發過程中,同時也產生了一定濃度的O3,進一步促進NOx的氧化,與YANG等[18]的研究相一致。在實驗過程中沒有引入水,反應裝置氣密性好,催化劑全部干燥。筆者認為反應過程中的水可能是催化劑或反應器表面吸附的水。圖3的XPS譜圖也支持了催化劑表面存在吸附水的可能性。在等離子體協同催化的過程中,CdS 的引入在一定程度上提高了等離子體放電的電場強度與能量密度從而提高了平均電子能量,使得CdS 更易受激發。
4 結論
在填充了γ-Al2O3載體的放電區域中引入了具有不同帶隙能的5 種催化劑,通過等離子體場中協同催化的綜合作用,提高了NO 和NOx的氧化效率,主要結論如下:1)N 型半導體催化劑WO3、ZnO、CdS的電子在電場中能夠被激活、選擇性獲取能量以參與協同催化反應,對于去除煙氣中的NOx表現出一定的能力;但P 型催化劑NiO 和Cu2O 的大部分載流子為空穴,在電場中被激活的幾率較小,獲得放電能量的機會較低,對協同催化過程影響小,氧化脫硝性能不理想;同一能量密度下,N型催化劑的帶隙能大小與氧化脫硝能力大小呈負相關。2)在40 kV放電電壓條件下,帶隙能最低的N 型催化劑CdS/γ-Al2O3對NO 與NOx的氧化率最高,與IC 檢測結果相吻合。
猜你喜歡
大自然探索(2023年7期)2023-11-14 13:08:06
石油石化綠色低碳(2019年6期)2019-02-13 09:39:01
石油石化綠色低碳(2019年6期)2019-01-14 01:16:22
智富時代(2018年3期)2018-06-11 16:10:44
浙江大學學報(工學版)(2016年11期)2016-06-05 09:21:04
Coco薇(2016年2期)2016-03-22 02:45:06
超硬材料工程(2016年1期)2016-02-28 22:20:04
中國資源綜合利用(2016年4期)2016-01-22 08:27:23
合成化學(2015年4期)2016-01-17 09:01:27
應用化工(2014年3期)2014-08-16 13:23:50
