六價硫氟交換化學在藥物設計中的應用*
2023-09-27 03:43:02桂馨墨秦藝曼
云南化工 2023年9期
張 煚,桂馨墨,秦藝曼
(炎癥免疫性疾病安徽省實驗室 安徽醫科大學藥學院,安徽 合肥 230032)
磺酰氟類化合物的合成及性質的研究最早可以追溯到20世紀20年代的德國,隨后,這些磺酰氟化合物被嘗試作為酶抑制劑。Gold和Fahrney在1963年首次介紹了磺酰氟對酯酶的抑制作用[1],并測定了不同結構的磺酰氟對乙酰膽堿酯酶、α-胰凝乳蛋白酶和胰蛋白酶的反應速率。20世紀,關于磺酰氟類酶抑制劑的研究,產生了眾多的成果。如今,細胞裂解液中為了防止蛋白降解而采用的蛋白酶抑制劑,如PMSF和AEBSF,即為磺酰氟類化合物。在早期的藥物開發研究中,Baker是研究芳基磺酰氟類共價抑制劑的先驅。早在20世紀60年代,Baker等就使用芳基磺酰氟類化合物作為二氫葉酸還原酶、胰蛋白酶、α-胰凝乳蛋白酶、鳥嘌呤脫氫酶和黃嘌呤氧化酶等多種酶的不可逆共價抑制劑[2-7]。Colman團隊亦在磺酰氟類共價抑制劑的研究中做了大量的工作[8-11]。但總的來說,長期以來,硫氟化合物在藥物設計領域沒有得到足夠的認識,關注度較低。
2014年,Scripps研究所的Sharpless(2001和2022年兩次諾貝爾化學獎得主)團隊首次提出六價硫氟交換化學(Sulfur(VI)fluoride exchange,SuFEx)是一類新型的點擊化學[12]。自此之后,六價硫氟交換化學引起了人們極大的關注,眾多重要的研究成果不斷涌現,已被廣泛應用于有機合成、材料化學、化學生物學和藥物化學等領域中[13]。鑒于六價硫氟交換化學在藥物分子的設計合成中日益增加的重要性,本文介紹了近年來SuFEx在藥物設計中的應用情況。
1 六價硫氟類化合物的結構及理化性質
目前,在藥物設計中得到廣泛應用的六價硫氟類化合物主要是烷基磺酰氟、芳基磺酰氟、芳基氧磺酰氟和芳基氟磺酰亞胺(圖1A)。相對于磺酰氯,磺酰氟具有更為惰性的化學反應活性,能耐受還原性條件,在生理pH的水溶液中具有較長的半衰期,具有相對較好的生物相容性,和親核試劑反應時只通過加成消除的取代機理形成共價鍵[14]。六價硫氟類彈頭與靶蛋白中的親核性氨基酸殘基反應,能夠形成穩定的共價復合物(圖1B),但在特殊的情況下,芳基氧磺酰氟與賴氨酸形成的共價鍵也會因蛋白介導的水解作用水解為氨基磺酸[15]。
(A)六價硫氟類化合物的結構;(B)六價硫氟化合物與靶蛋白反應形成共價復合物。圖1 六價硫氟類化合物
為了更好地設計含六價硫氟彈頭的共價抑制劑,Mukherjee等詳細考察了烷基磺酰氟、芳基磺酰氟、乙烯基磺酰氟、芳基氧磺酰氟和芳基氟磺酰亞胺等六價硫氟化合物(圖1A)在生理條件下與親核性氨基酸殘基的反應活性[15],結果表明:烷基磺酰氟的反應活性最高,穩定性最差;乙烯基磺酰氟和芳基氟磺酰亞胺的穩定性高于芳基磺酰氟且活性與取代基相關;芳基氧磺酰氟最為惰性,反應活性最差,十分穩定;芳基磺酰氟的反應活性和生理條件下的水解性均與芳環上取代基直接相關,吸電子的取代基大幅提高活性的同時降低其水解穩定性。因此,在藥物化學中,設計共價抑制劑時可以據此優化目標化合物的結構,調節反應活性與選擇性,以達到恰當的平衡。
2 含六價硫氟官能團的小分子抑制劑
近年來,共價抑制劑的研究得到了蓬勃發展。與非共價藥物相比,共價鍵的引入可以提高藥物的活性,延長藥物在體內的駐留時間等。對那些難以成藥的靶點來說,共價藥物是個很好的選擇。在傳統的共價藥物設計時,通常借助于α,β-不飽和酰胺(酮)等結構[16],但共價藥物的化學選擇性,以及伴隨而來的脫靶效應一直困擾著我們,遲滯了共價藥物的開發。六價硫氟類(SuFEx)彈頭,尤其是芳基氧磺酰氟,在生命體系中能夠恰當地平衡反應活性和選擇性。與靶標蛋白結合后,基于臨近效應,能夠高度選擇性地與特定的氨基酸殘基(Tyr、Ser、Thr 和Lys 等)反應,形成穩定的共價復合物;而與蛋白組的反應性很低,脫靶效應很弱。因此,含有SuFEx彈頭的化合物具有作為共價藥物的巨大潛力。幾種典型的含六價硫氟官能團的小分子抑制劑案例如下文所示。
泛素蛋白酶體系統(UPS),是真核細胞內蛋白質降解的主要途徑,負責降解錯誤折疊、損傷的非正常蛋白質,對維持細胞正常生理狀態具有十分重要的作用。同時,它也是抗腫瘤靶點,目前已有硼替佐米、卡非佐米等蛋白酶體抑制劑被批準上市,用于治療多發性骨髓瘤等惡性腫瘤。鑒于磺酰氟獨特的化學性質,Brouwer等設計合成了一系列氨基酸磺酰氟衍生物[17-18],并基于此開發了選擇性高且活性好的擬肽類磺酰氟蛋白酶體抑制劑(Peptido Sulfonyl Fluorides,PSFs)[19-24](圖2)。化合物6對絲氨酸蛋白酶具有一定的抑制作用,其對糜蛋白酶的Ki值為 22 μmol/L。化合物7對酵母20S蛋白酶體的IC50達到 7 nmol/L。化合物8對蛋白酶體的抑制作用稍弱,但具有一定的選擇性,對蛋白酶體β5i亞基的抑制作用比β5c亞基高25倍。進一步的修飾發現,具有游離氨基的擬肽類蛋白酶體抑制劑9-11具有納摩爾級的IC50值,并且在不同的蛋白酶體亞基間具有顯著的選擇性。

圖2 擬肽類磺酰氟抑制劑的結構及活性
轉甲狀腺蛋白(transthyretin,TTR),一種以四聚體形式轉運甲狀腺素和視黃醇的重要蛋白。在病理情況下,其解聚為單體,并錯誤折疊或組裝生成淀粉樣纖維,從而導致多種淀粉樣蛋白集聚相關的疾病。Kelly等合成了兩個芳基磺酰氟(12,13)和兩個芳基氧磺酰氟(14,15)取代的1,3,4-噁二唑類TTR共價穩定劑(圖3)[24-25]。該類化合物可以共價地與甲狀腺素結合口袋中Lys15共價結合,形成磺酰胺鍵,從而穩定TTR。其中,芳基氧磺酰氟類化合物14和15由于活性較低,與Lys15的反應較慢,且不能反應完全,但對TTR的選擇性更高,具有較弱的脫靶效應。此外,其與TTR形成的共價物顯示出很強的熒光性,可以用來在活細胞和秀麗隱桿線蟲體內檢測TTR。一個有趣的現象是,化合物14和15與TTR中Lys15形成的芳基氧磺酰胺共價物會因蛋白結構的誘導作用水解為TTR-SO3。

圖3 六價硫氟類TTR共價穩定劑
抗生素耐藥是當前嚴重威脅人類健康的因素之一,為了克服細菌的多藥耐藥,筆者在Sharpless教授指導下基于SuFEx化學,由酚或酚的前體制備芳基氧磺酰氟衍生物,通過抗菌活性的表型篩選,發現白藜蘆醇、己雷瑣辛與和厚樸酚的芳基氧磺酰氟衍生物(16-18)(圖4)可以有效殺死MRSA、VRSA、VRE和多種臨床分離得到的多藥耐藥菌。這些衍生物沒有明顯的細胞毒性,能夠高效破壞細菌的生物膜且不易產生耐藥菌株,己雷瑣辛的衍生物還與抗菌藥物鏈霉素和慶大霉素具有協同效應[26]。

圖4 具有抗耐藥菌活性的白藜蘆醇、己雷瑣辛與和厚樸酚的芳基氧磺酰氟衍生物
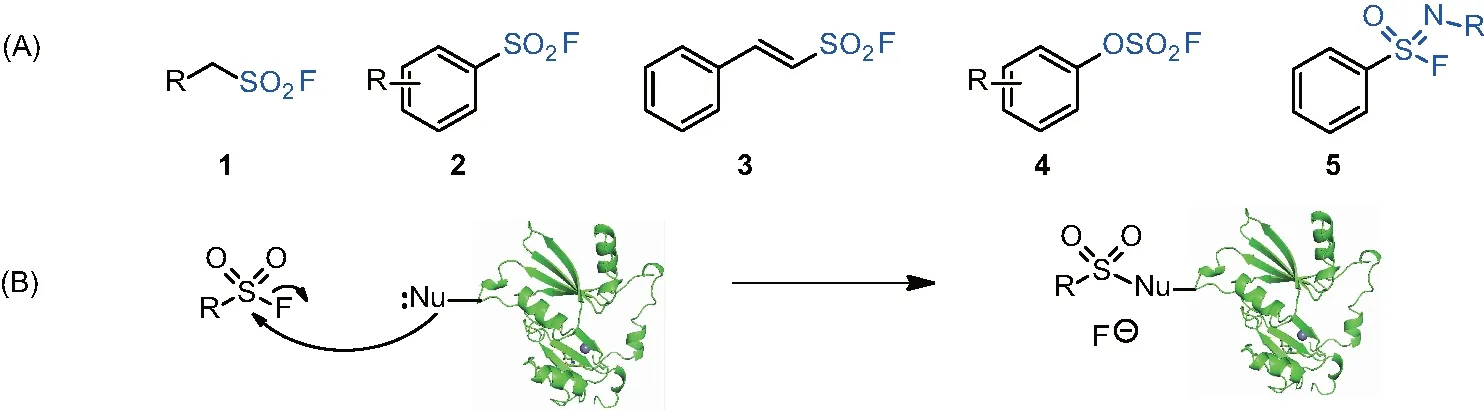
常規的藥物發現策略是通過篩選或合理藥物設計來獲得靶向特定蛋白的小分子。Kelly團隊開發了一種逆向藥物發現方法學,即通過溫和反應性的小分子來識別蛋白質組中的親核位點,再進一步有針對性地優化先導物獲得潛在藥物分子。SuFEx類化合物在生物體內的反應性和穩定性間具有良好的平衡,適合在逆向藥物發現策略中作為探針。Kelly團隊制備了芳基氟磺酰亞胺類小分子化合物庫,并進一步鑒定這些SuFEx化合物所共價結合的蛋白質。其中,基于胸苷分子的芳基氟磺酰亞胺19(圖5)可以共價結合多聚ADP核糖轉移酶1(poly ADP-ribose polymerase 1,PARP1)并抑制其活性,且在活細胞中也具有良好的活性。基于此開發的藥物分子有潛力作為治療癌癥的PARP1非共價抑制劑的有效補充[27]。
(A)芳基氟磺酰亞胺類衍生物的合成;(B)基于胸苷分子的芳基氟磺酰亞胺衍生物19。圖5 逆向藥物發現策略考察芳基氟磺酰亞胺類的作用靶點
3 基于SuFEx的化合物庫構建
SuFEx類官能團的化學穩定性高,但在合適的反應條件下針對特定官能團又具有十分高效的反應活性,選擇性高,因此可以用來模塊化地構建衍生物庫,以供生物活性篩選。例如,很多臨床使用的藥物分子中含有酚羥基,而硫酰氟(SO2F2)可以高度選擇性地修飾酚羥基。Liu等采用后期官能團化(Late-Stage Functionalization,LSF)的衍生物合成策略,用SO2F2氣體的乙腈溶液在96孔板中一次性將39個含酚羥基的抗癌藥物原位轉化為芳基氧磺酰氟衍生物,測試發現相對母體化合物,氧磺酰氟標記的衍生物20F-22F(圖6)可以大幅提高抗癌活性[28]。

圖6 芳基氧磺酰氟類抗癌藥物
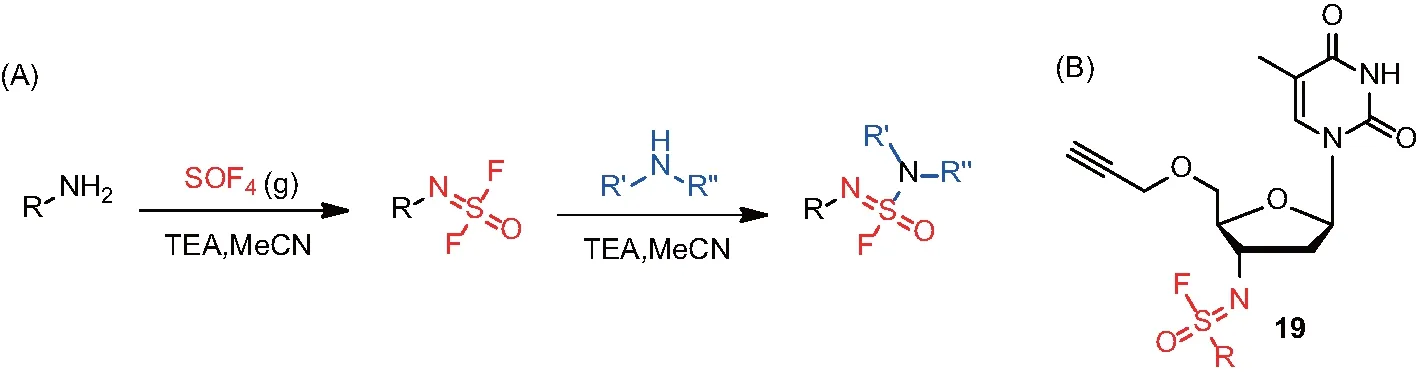
針對苗頭化合物進行結構優化,最終獲得藥物分子的過程十分漫長且充滿挑戰性。SuFEx點擊化學具有生物相容性,基于此構建的衍生物庫可以直接用于原位的生物活性篩選。Wolan團隊和Sharpless團隊合作報道了一種基于SuFEx點擊化學快速制備芳基氟磺酰亞胺衍生物庫的策略[29];研究者從細菌半胱氨酸蛋白酶SpeB的中等活性抑制劑(23,Ki=8 μmol/L)出發,通過SuFEx點擊化學策略在微孔板中快速(過夜)合成460種衍生物(圖7A),通過直接原位篩選發現了多個活性得以大幅提高的衍生物。其中,活性最高的衍生物Ki為 18 nmol/L,衍生物24活性較好(Ki為 67 nmol/L),水溶性好,細胞毒性低,代謝穩定性高,成藥性好(圖7B)。
圖7 基于SuFEx點擊化學構建衍生物庫發現細菌半胱氨酸蛋白酶SpeB抑制劑
4 基于SuFEx的共價蛋白藥物

圖8 非天然氨基酸FSY的結構及SuFEx介導的蛋白質偶聯
5 基于18F/19F同位素交換的PET示蹤劑的合成
基于18F的正電子發射斷層顯像(Positron Emission Tomography,PET)技術在疾病的診斷和病理研究等方面具有十分廣泛的應用。未來這一技術的主要研究方向之一是在18F脫氧葡糖(18F-FDG)等示蹤劑之外,開發更多具有靶向性的18F標記的示蹤劑,以滿足日益增長的精準醫療的需求。由于18F的半衰期較短,只有 110 min,因此在示蹤劑中通過常見的碳-氟鍵引入18F,并在有限時間里快速分離純化,非常具有挑戰性。Scripps研究所Wu Peng團隊證實了芳基氧磺酰酯在室溫下可以快速與無機氟鹽發生18F/19F同位素交換,并基于此開發了一種基于硫氟交換的超快18F標記技術[35]。這一技術得到的18F標記的示蹤劑具有高比活度和高放化純度。研究者應用該項技術在 2 min 之內便近乎當量地獲得18F標記的PARP抑制劑奧拉帕尼,并且在小鼠的PET/CT中證實其對PARP高表達的腫瘤組織具有較高的選擇性。基于該技術,He等合成了新型的阿爾茨海默病中β-淀粉樣蛋白斑塊(Aβ)的PET放射性示蹤劑[36]。為進一步提高基于SuFEx的18F標記效率,Walter等開發了一種新的方案[37],即將[18F]F-負載到陰離子交換樹脂上,并用BnEt3NCl的MeOH溶液洗脫,以獲得18F標記的示蹤劑。這種方法使得我們能夠在高度稀釋的溶液中對低納摩爾量的芳基氧磺酰氟進行18F標記。和最初的方案相比,該方法避免了共沸干燥、堿的添加和HPLC純化。作者利用這一方式,成功制備了29種18F-芳基氧磺酰氟,并且初步評估表明[18F]FS-DPA是一種非常有前景的易位蛋白(TSPO)表達可視化的示蹤劑。Kim等[38]近來也開發了一種18F標記方案,即通過[18F]FSO2+轉移試劑,可以原位地由苯酚或者胺制備芳基氧磺酰[18F]氟或氨基磺酰[18F]氟(圖9)。
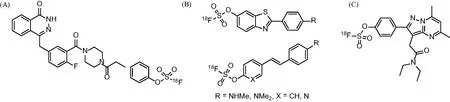
(A)18F標記的奧拉帕尼芳基氧磺酰氟衍生物;(B)18F標記的β-淀粉樣蛋白斑塊(Aβ)的PET示蹤劑;(C)[18F]FS-DPA。圖9 18F標記的PET示蹤劑
6 展望
六價硫氟類化合物具有特殊的理化性質,在化學穩定性和反應性上具有很好的平衡。近年來,隨著SuFEx的新型合成砌塊、新型合成技術的不斷涌現,藥物化學家擁有了更多的工具來修飾小分子和生物大分子為六價硫氟衍生物,并基于此發現共價小分子和大分子藥物。但當前六價硫氟化合物的反應機制尚未完全明確,該類共價抑制劑的脫靶效應,尤其是體內的毒副作用還需進一步研究。未來,隨著研究的不斷深入,六價硫氟交換化學在藥物的設計領域具有廣闊的應用前景。
