動物源性醫(yī)療器械中殘留DNA表征和風險的研究進展
2023-12-08 14:36:48孫曉霞劉成虎朱藝馨屈秋錦阮文婷
科學技術與工程 2023年31期
關鍵詞:檢測
孫曉霞, 劉成虎, 朱藝馨, 屈秋錦, 阮文婷
(山東省醫(yī)療器械和藥品包裝檢驗研究院, 國家藥品監(jiān)督管理局生物材料安全性評價重點實驗室, 濟南 250101)
隨著生物醫(yī)藥、工程材料等新技術的飛速發(fā)展,動物源性醫(yī)療器械被越來越多地用于疾病治療,以實現(xiàn)修復、替代、維持或改善組織和器官功能的目的。這是因為,相較于金屬、合成高分子等人工合成材料,動物源性醫(yī)療器械不僅具有良好的生物相容性,其廣泛的組織來源還能提供類似于人體的三維網(wǎng)狀支架結構以利于組織再生修復[1-2]。為了降低針對動物源性成分的免疫風險,在醫(yī)療器械的生產(chǎn)過程中常采用脫細胞等工藝,去除細胞等相關免疫原性成分[3-4],但由于DNA具有穩(wěn)定性,其終產(chǎn)品上仍有可能殘留宿主細胞DNA[5-6],因此早在1987年世界衛(wèi)生組織(World Health Organization,WHO)就將殘留DNA含量作為動物源性醫(yī)療器械質量控制的一項關鍵性能指標[7]。
許多研究顯示動物源性材料中殘留的DNA具有生物活性,尤其當動物源性材料中含有病毒感染細胞或致癌/致瘤潛能的細胞時,其DNA亦可能攜帶HIV病毒或Ras癌基因等片段[8-10],從而導致病毒感染、致瘤和異常基因表達等安全性風險。同時,風險評估研究認為,DNA片段大小的減小極有可能進一步降低DNA的風險并增加安全裕度,因為殘留的DNA片段越小,存在完整癌基因和其他功能序列的可能性就越低[11-13]。由此可見,采用適宜檢測方法表征的殘留DNA水平對于保證動物源性醫(yī)療器械的安全性和質量可控性至關重要。
由于具有學科綜合性強、技術含量和附加值高的特點,使動物源性醫(yī)療器械產(chǎn)品的研發(fā)成為熱點。盡管目前關于動物源性醫(yī)療器械中可接受殘留DNA水平(即DNA限值)的研究仍較少,但國內外各類監(jiān)管機構已發(fā)布了細胞制品等生物源性材料的指導原則和建議要求[14-16]。隨著DNA風險研究的深入,監(jiān)管機構也在不斷更新優(yōu)化殘留DNA限值要求,這將有助于確保動物源性等生物材料被科學管理、安全使用。此外,聚合酶鏈反應(polymerase chain reaction, PCR)等分子生物學技術的發(fā)展,使殘留DNA的檢測方法更加科學、準確,為動物源性醫(yī)療器械產(chǎn)品開展合理的質量控制提供了技術保證[17-19]。通過分析動物源性材料中殘留DNA的潛在風險,回顧和比較監(jiān)管機構已發(fā)布的生物制品中殘留DNA的限值要求和各國藥典中收錄的殘留DNA測定方法;同時結合工藝、材料控制等充分考慮風險,探討降低DNA殘留量的措施手段。旨在推動動物源性醫(yī)療器械生產(chǎn)技術水平提高,有效去除器械中殘留DNA,通過合理的質量控制,正確進行風險/收益評估,從而保障動物源性醫(yī)療器械的安全使用。
1 殘留DNA的風險來源和潛在危害
DNA是遺傳物質的載體,存儲著生物體的形態(tài)和功能信息。通過受體介導的內吞作用,機體代謝產(chǎn)生的凋亡細胞DNA和進入機體的外源性DNA能夠被哺乳動物細胞攝取,由細胞內核酸酶降解和甲基化酶甲基化而失活,隨著細胞分裂被逐漸清理[20-21]。而未被降解的外源性DNA,特別是一些小片段DNA片段(長度約500 bp)則可能進入細胞核、插入基因組[22-23],由此產(chǎn)生的生物活性作用將導致不可預見的安全性風險。由于不同來源的動物源性材料具有不同的潛在風險,并且殘留DNA片段大小各異,使其風險復雜多樣。1999年美國食品藥品監(jiān)督管理局(U.S. Food and Drug Administration, FDA)在關于“細胞來源疫苗的監(jiān)管進展”工作組會上,回顧以往生物材料使用史,指出宿主細胞中殘留DNA具有致癌性、感染性、免疫原性以及致突變性等風險[24]。
1.1 致瘤性/致癌性
殘留DNA致癌的主要機制是引入了顯性致癌基因。如果編碼癌基因的殘留DNA進入細胞核病并激活相應的癌基因,正常細胞會發(fā)生轉化并分化為瘤細胞。Zheng等[12]、Sheng等[25]的研究已證實表達癌基因H-ras和c-myc的質粒DNA可以在皮下接種后誘導含有這兩種癌蛋白的腫瘤形成,并且1 μg的質粒DNA即可誘導新生NIH Swiss小鼠成瘤[9]。
此外,在禽類和哺乳動物中還觀察到由于殘留DNA插入導致的腫瘤形成[26-30]。然而,由于不同種屬間DNA序列的異質性和基因組復雜性[31-32],使得這種插入后發(fā)生DNA整合并且整合位置在原癌基因或抑癌基因區(qū)域的概率非常低,其中整合并活化一個癌基因的概率是10-10,而原癌基因激活和抑癌基因失活同時發(fā)生的概率是10-19[33-34]。因此,相對于插入引起的致癌性,人們更需要關注殘留DNA引入的顯性致癌基因。對殘留DNA的致癌性研究提示人們在選擇動物源性材料時,應盡量選取正常的二倍體細胞,避免采用連續(xù)傳代或具有致癌潛能的細胞,同時也要控制DNA殘留量和DNA片段大小,從而盡可能地降低殘留DNA的致癌性風險。
1.2 感染性
殘留DNA的感染性風險來自于細胞中存在的感染性病毒基因組。研究發(fā)現(xiàn)許多病毒基因組具有感染性,例如人乳頭瘤病毒(HPV)[35]、腺病毒[36]、皰疹病毒[37]、EB病毒(EBV)[38]、乙肝病毒(HBV)[39]等,通過將病毒DNA整合到宿主細胞基因組上而產(chǎn)生感染性病毒。除了DNA病毒,外源性逆轉錄病毒也具有感染性風險,如來自鳥類和嚙齒動物的逆轉錄病毒(迄今為止還未發(fā)現(xiàn)人類內源性的逆轉錄病毒具有感染性)。由于宿主細胞中存在的多種感染性病毒基因組,可見DNA感染性風險可能比致癌性風險更高。Sheng-Fowler等[8]、Chabot等[40]對HIV病毒DNA的研究表明,線狀DNA的感染力是環(huán)狀DNA的30~100倍,1 pg殘留線狀DNA就具有感染性,而HIV感染細胞中環(huán)狀DNA的致病劑量為2 μg。考慮到殘留DNA的病毒感染性風險,除了對動物源性材料中病毒進行滅活,人們還需要從殘留DNA的數(shù)量和片段方面來降低病毒感染性。
1.3 免疫原性
盡管DNA分子缺乏固定的空間結構、不能作為抗原被抗體識別,然而當DNA被設計成編碼某種蛋白質抗原的重組真核表達載體,即作為DNA疫苗,外源基因可以在體內表達,產(chǎn)生的抗原能夠激活機體免疫系統(tǒng)而用于免疫治療[41]。但許多研究報道,DNA免疫時可在注射局部引起非治療效應的炎癥反應[41-42]。這可能與DNA誘導局部免疫應答而介導的免疫刺激作用有關,例如根據(jù)Hornung等[43]的研究結果,細胞中的DNA能夠與酶結合形成復合物而激活天然免疫系統(tǒng),釋放炎癥小體引起發(fā)熱和局部腫脹等炎癥癥狀。此外,Mostafa等[44]研究發(fā)現(xiàn)自閉癥兒童的血液中抗雙鏈DNA(ds-DNA)抗體陽性率(34%)明顯高于健康兒童(2%),提示這些抗體可能參與自閉癥兒童的自身免疫攻擊。若動物源性材料中殘留的DNA與兒童自身的DNA序列相似或重疊,盡管這種情況的發(fā)生概率極低,但當兒童暴露于這種非自身DNA片段時,則可能會產(chǎn)生與其自身DNA相交叉的免疫反應,誘發(fā)機體免疫損傷。
1.4 致突變性
DNA插入基因組后還可能導致基因突變。研究發(fā)現(xiàn)X1-連鎖嚴重聯(lián)合免疫缺陷病(SCID-X1)患者接受基因治療后,常發(fā)生基因易位和突變等異常而導致治療失敗[45-47],提示外源性DNA片段可能被外周血干細胞攝取、引起基因突變。當突變發(fā)生于膠質細胞時,則可能導致神經(jīng)系統(tǒng)疾病。例如,DNA雙鏈斷裂(double strand break, DSB)不僅見于紫外線、電離輻射以及烷化劑等化學性損傷,還是神經(jīng)活動中一個關鍵且正常的過程并能被機體快速修復[48]。研究顯示,參與DSB修復的基因在自閉癥中發(fā)生了突變,導致錯誤的基因重組或修復失敗[49-50]。此外近來研究發(fā)現(xiàn),精神分裂癥等神經(jīng)發(fā)育障礙性疾病與保護性基因缺失或異常導致的γ-氨基丁酸(γ-aminobutyric acid,GABA)能信號系統(tǒng)功能失調密切相關[51]。這些由DNA插入誘發(fā)的基因突變研究提示,盡管外源性DNA插入基因組并在特點位點整合的可能性很低[52-54],但對于本就具有基礎性疾病的患者或DNA代謝活躍的人群,由外源性DNA插入導致的基因突變風險將極大增加,引起基因重組異常或修復失敗以及多個組織器官功能障礙。
2 關于殘留DNA的法規(guī)要求
動物源性材料作為一個快速發(fā)展的新興技術領域,目前尚無此類生物材料中殘留DNA規(guī)定和要求,有關殘留DNA的基本法規(guī)均來自疫苗等生物制品。在生物制品的生產(chǎn)過程中,由于細胞基質殘留的DNA可能存在致癌性、感染性等安全性問題,因此各監(jiān)管機構(WHO、FDA及NMPA等)對殘留DNA進行了嚴格把控,以保證生物制品的安全使用。這里匯總了過去數(shù)十年,國內外監(jiān)管機構出臺的生物制品中可接受的殘留DNA水平的法規(guī)要求。
2.1 國外
細胞等生物材料中殘留DNA的安全性風險主要與其潛在致癌性有關,這是由于DNA插入基因組后可能發(fā)生癌基因激活。但最初由于研究數(shù)據(jù)有限,世界衛(wèi)生組織(WHO)根據(jù)Vero細胞來源的脊髓灰質炎疫苗中DNA含量,采用10 pg/劑作為疫苗中殘留DNA限量[55]。之后,隨著致癌性相關實驗數(shù)據(jù)的增多,研究人員假設在體內1 ng DNA對應的活化癌基因約為100個拷貝,由致癌性風險評估得出機體發(fā)生致癌性轉化的概率為10-9[56]。由此WHO的生物制品工作組推斷,100 pg DNA約對應1個拷貝的活化癌基因,發(fā)生致癌性轉化的概率為0.5×10-10且該風險是可以忽略的,于是在1986年WHO將殘留DNA的限值提高為100 pg/劑[7, 57]。然而該限值仍較嚴格,尤其是對于某些病毒疫苗,例如某些包膜病毒常常難以符合限值要求,由此導致相關疫苗的生產(chǎn)成本增加、價格上漲甚至造成嚴重的市場短缺。進一步的癌癥研究發(fā)現(xiàn),DNA誘導致癌還與基因突變、表觀遺傳等多種機制密切相關。此外,對于連續(xù)傳代的非致癌細胞,如常見的Vero細胞等,這些細胞中不太可能存在活化的顯性癌基因[58-60]。因此WHO在1997年組織專家再次討論并修改了DNA的限值,并認為減小殘留DNA片段大小有可能進一步降低致癌性、感染性等DNA的風險[61]。
目前WHO、美國FDA和歐洲藥典建議,生物材料的風險評估時應考慮原料細胞的性質和給藥途徑。WHO和美國FDA推薦,來源于連續(xù)傳代非致瘤細胞的生物制品,當胃腸道外使用時,無論是否用于嬰幼兒,其殘留DNA應不高于10 ng/劑,長度不大于200 bp[24, 62-63]。同時,基于DNA經(jīng)口和胃腸道暴露的風險差異,FDA建議將口服類疫苗的DNA殘留量放寬至100 μg/劑;以人二倍體細胞系來源(如被廣泛用于生物制品的MRC-5細胞和WI-38細胞)的生物制品,FDA不認為其殘留DNA具有安全性風險[64]。相對于WHO,歐盟提高了嬰幼兒使用疫苗的要求,嬰幼兒疫苗≤100 pg/劑,非嬰幼兒疫苗≤10 ng/劑。歐洲藥典10.0根據(jù)最小的功能片段長度,認為<200 bp的DNA是安全的[16]。
2.2 中國
中國藥典2020年版三部規(guī)定,以大腸桿菌或真菌基質生產(chǎn)的疫苗和治療性生物制品,其DNA殘留量不能超過10 ng/劑;對于以細胞基質生產(chǎn)的滅活疫苗和治療性生物制品,其DNA殘留量應低于100 pg/劑[14]。同時,藥典2020年版還優(yōu)化了安全性檢測方法,除了DNA探針雜交法和熒光染色法,在通則3407“外源性DNA殘留量測定法”中新增了“定量PCR法”,通過進一步完善生物材料的質量控制方法,將人用狂犬病疫苗的DNA殘留量由100 pg/劑調整為3 ng/劑,然而以CHO細胞為來源的重組乙型肝炎疫苗仍保留10 pg/劑的限值要求[17]。中國現(xiàn)行2020版藥典規(guī)定檢測DNA殘留量的疫苗、生物制品及標準匯總分別如表1(疫苗)和表2(生物制品)所示[14]。
3 殘留DNA的測定方法
基于動物源性醫(yī)療器械的安全性和質量可控性,建立準確、靈敏且特異性強的殘留DNA測定方法是至關重要的。各國藥典給出了數(shù)種檢測方法用于表征殘留DNA,包括DNA探針雜交法、熒光染色法定量PCR法以及基于DNA結合蛋白免疫酶法[15-17, 65-66]。這些方法可用于在產(chǎn)品生產(chǎn)過程中和成品上測定DNA殘留量和DNA片段大小。
3.1 DNA探針雜交法
供試品中的DNA經(jīng)變性為單鏈后吸附于固相膜上,在一定條件下(適宜的溫度及離子強度等)與特異性標記的單鏈DNA探針退火復性、雜交形成雙鏈DNA,使用與標記物相應的檢測系統(tǒng)顯示雜交結果,顏色深度與DNA含量相對應,通過與已知含量的陽性DNA比對,計算供試品中的DNA殘留量[15, 17]。DNA探針雜交技術,將已知的DNA 序列片段作為雜交探針,通過放射性或熒光標記,能夠特異性檢測未知的DNA樣品片段及其相對大小。然而在實際操作時,放射性標記物(如32P)存在半衰期短、放射等問題,而熒光標記的探針如果采用儀器讀取信號,雜交法理論上可以達到定量檢測要求的靈敏度,但是檢測時間需要48 h;由此可見,這種方法的操作比較煩瑣、費時,操作過程中殘留的溶劑可能會干擾檢測結果,這些不足限制了雜交法的應用。
3.2 免疫閾值法
供試品中DNA變性為單鏈DNA(ssDNA)后,能夠同時與鏈霉親和素標記的ssDNA結合蛋白和脲酶偶聯(lián)的抗ssDNA單抗結合,形成含有DNA-鏈霉親和素-脲酶的復合物并被生物素化膜捕獲。在尿素溶液中,膜上的復合物通過與尿素反應產(chǎn)生氨(NH3),引起溶液pH改變且改變與DNA含量成正比[15-16]。因為是采用DNA抗體的非特異序列免疫檢測技術,因此閾值法不能特異性識別宿主殘留DNA序列,且容易受到環(huán)境和操作人員的DNA污染,導致讀值偏高。
3.3 定量PCR法(qPCR法)
針對供試品DNA設計特異性引物和帶有熒光標記的探針,在PCR反應過程中,通過熒光標記探針或摻入熒光染料,連續(xù)監(jiān)測反應體系中熒光數(shù)值的變化,實時反映特異性擴增產(chǎn)物量的變化。當PCR反應釋放的熒光強度達到預設的閾值時,體系的PCR循環(huán)數(shù)(Ct值)與該體系所含的起始DNA模板量的對數(shù)值呈線性關系;采用已知含量的DNA標準品構建標準曲線,通過Ct值計算供試品中的DNA殘留量[15-17]。qPCR法具有序列特異性,其靈敏度、準確度、精密度等性能好,可以實現(xiàn)快速、高通量DNA片段檢測;其中,特異且高度保守的q-PCR引物/探針等試劑是保證殘留DNA準確可靠檢測的關鍵。

表2 中國藥典規(guī)定的治療性生物制品中DNA殘留量標準[14]Table 2 The limits of residual DNA in therapeutic biologics according to provisions for Chinese Pharmacopoeia[14]
3.4 熒光染色法
這種方法僅適用于測定雙鏈DNA。雙鏈DNA熒光染料(PicoGreen)與供試品中的雙鏈DNA特異結合形成復合物,在波長480 nm激發(fā)下產(chǎn)生熒光信號,使用熒光酶標儀在波長520 nm處進行檢測;在一定DNA濃度范圍內,熒光強度與DNA濃度成正比,可采用已知含量的雙鏈DNA標準品構建標準曲線,根據(jù)熒光強度計算供試品中的DNA殘留量[17]。除了雙鏈DNA,動物源性醫(yī)療器械中還可能殘留單鏈DNA片段,且殘留的DNA片段大小不一,而這種方法中僅僅是將已知含量的雙鏈DNA作為標準品,這些極大的影響了殘留DNA檢測的準確性。
在選擇合適的殘留DNA檢測方法時,需結合樣品的特征考慮檢測方法的特異性和局限性(見表3)。其中,qPCR方法同時被中國藥典[17]、美國藥典[15]和歐洲藥典[16]收錄,其技術優(yōu)勢在于序列特異性高、靈敏度高、重現(xiàn)性好,還可以同時實現(xiàn)定量和相對分子質量的高通量檢測,使得操作更簡便、結果更精確,從而為企業(yè)在工藝研究和成品質量控制方面提供了可靠的檢測手段。而現(xiàn)行版中國藥典收錄的以PicoGreen為代表的熒光染料法[17],由于存在技術缺陷、不能準確定量,已經(jīng)被美國和歐盟藥典摒棄。
4 降低DNA風險的手段
對于動物源性醫(yī)療器械中殘留DNA的風險,控制其DNA生物活性是關鍵。除了減少DNA殘留量,對于胃腸外使用的生物制品,由于殘留的DNA中可能包含致癌基因,WHO還建議將DNA片段化,使其大小降低到功能基因以下,即小于200個堿基對[63]。目前常用的降低DNA殘留量及其大小的方法包括的酶解、滲濾、超濾、層析等[67-69]。在生物材料中加入核酸酶,如DNase Ⅰ、非限制性核酸內切酶等,可以靶向DNA將大分子酶解成小片段并降低了其分子黏度,但這種方式產(chǎn)生的DNA片段大小不一,還會導致酶的殘留。根據(jù)DNA帶有負電荷的特點,可使用富含正電荷的物質,如聚乙烯亞胺(polyethyleneimine, PEI)、魚精蛋白等與之中和形成沉淀而去除DNA[70];也可以采用陰離子交換劑或親和層析的方式,通過控制吸附、洗脫條件來除去帶負電荷的DNA[71]。盡管利用電荷來去除DNA的操作簡便,但不適用于同樣帶有負電荷的材料;若產(chǎn)品帶正電荷,還會與DNA發(fā)生聚集和包裹,導致DNA去除效果不理想[72]。此外,也可采取化學滅活的方式,如β-丙內酯(BPL)等,降低產(chǎn)品中病毒DNA的大小和活性。
然而動物源性醫(yī)療器械生產(chǎn)過程中的一般工藝難以有效去除DNA,這一方面是由于DNA超強的化學穩(wěn)定性,另一方面是由于DNA呈負電荷易與其他生物大分子結合從而產(chǎn)生聚集(或吸附)、包裹作用。考慮到不同的組織來源和用途,可以通過多種去除方式組合的方案,以實現(xiàn)更加充分的DNA去除效果。
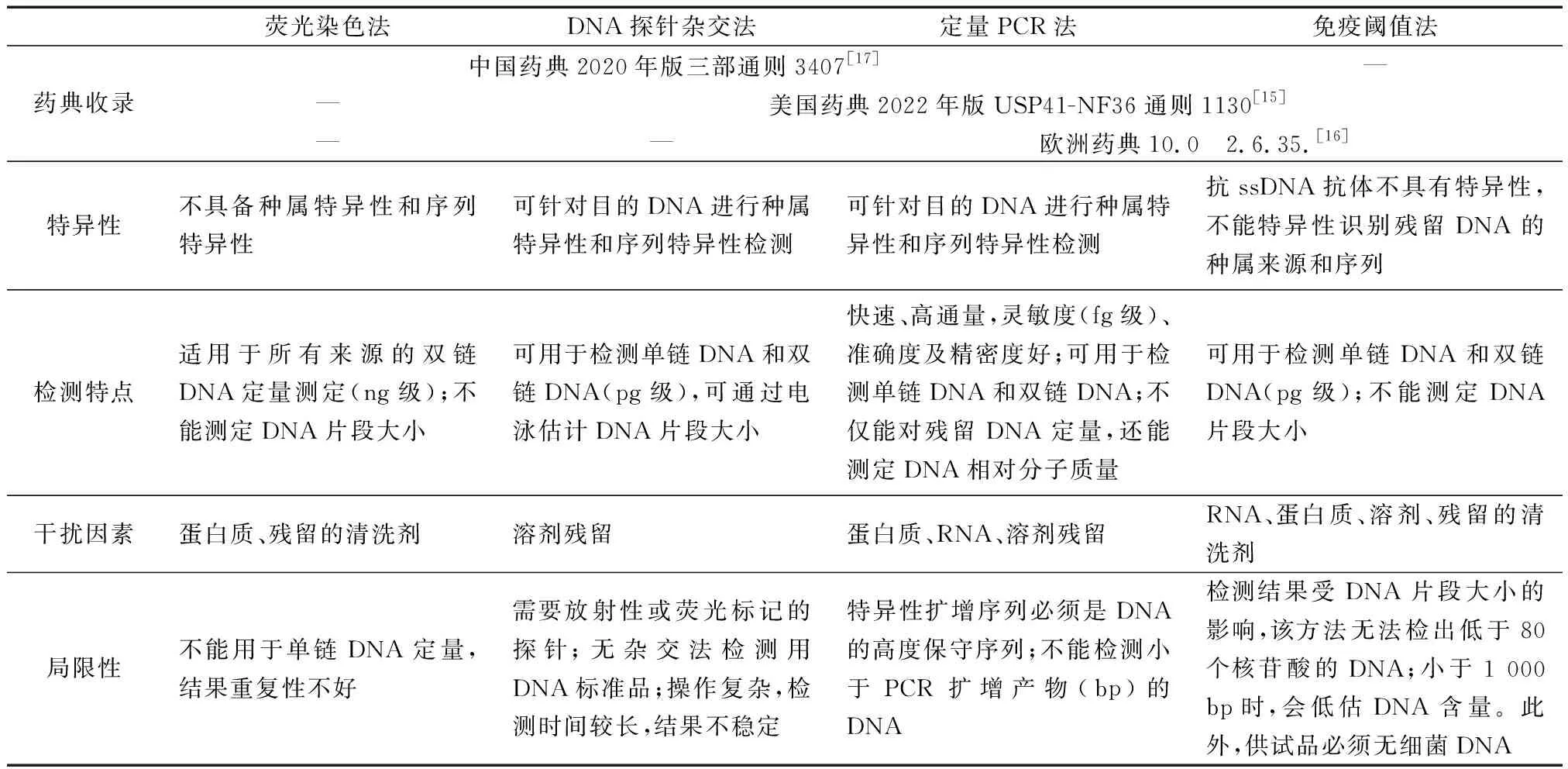
表3 不同DNA殘留量檢測方法異同Table 3 Comparison of the methods for residual DNA testing
5 總結與展望
隨著越來越多的動物源性醫(yī)療器械參與疾病治療,對其質量控制也日益嚴格。對于去除免疫原性物質的醫(yī)療器械,盡管在制造過程中進行了脫細胞等工藝,但最終產(chǎn)品中仍可能殘留DNA片段。因此,動物源性醫(yī)療器械中殘留的DNA同樣存在安全性風險和需要開展風險管理。
基于風險分析的安全性研究是進行產(chǎn)品質量控制的基礎,通過分析動物細胞殘留DNA的潛在風險和來源,追溯了生物制品中殘留DNA可接受水平的演變,顯示該限值是基于不同材料來源和使用條件下的殘留DNA風險評估過程。盡管相較于生物制品,動物源性醫(yī)療器械與人體的接觸方式更多樣,如表面接觸、外部接入、植入等[73],但它們都來自于生物材料且不可避免的殘留了宿主細胞DNA,這提示針對動物源性醫(yī)療器械中殘留DNA開展風險管理時,除了分析這些DNA可能編碼或隱藏的癌基因或感染因子等潛在危害,還應關注不同類型的醫(yī)療器械,通過深入探究不同接觸方式、接觸周期器械的安全性問題,為科學監(jiān)管提供殘留DNA表征和穩(wěn)健風險評估等數(shù)據(jù)支持,從而保障此類器械的安全使用。
為了盡可能去除外源污染、確保產(chǎn)品質量,對宿主細胞殘留DNA的表征和測試是控制動物源性醫(yī)療器械生產(chǎn)的一個關鍵組成部分。這些殘留DNA的生物活性決定了其安全性風險,它與DNA的多少和片段大小密切相關。FDA等的監(jiān)管指南建議胃腸外使用生物制品中殘留DNA水平為10 ng/劑和200個堿基對大小,而中國藥典僅對殘留量的限值進行了要求(如表1和表2所示)。由于DNA片段大小與其功能活性密切相關[8-10, 22-23],并且風險評估的研究表明,相同質量DNA的條件下,DNA片段的減小將有助于降低風險[11-12]。這說明除了DNA含量,在動物源性醫(yī)療器械產(chǎn)品進行質量控制、提高安全性時,還應關注殘留的DNA片段大小。
殘留DNA表征的目的包括:①確認純化工藝合理,能有效去除宿主DNA殘余;②確認產(chǎn)品中雜質含量符合標準要求。非特異性的DNA檢測結果不能區(qū)分究竟是生產(chǎn)中污染、檢測污染、還是工藝缺陷引起的DNA殘留,所以無法為解決方案提供有效信息。在嚴格的生產(chǎn)體系中,殘留DNA準確表征能夠解決工藝合理性問題。為了加強對產(chǎn)品生產(chǎn)的過程監(jiān)測和質量控制,各國藥典都對殘留DNA的檢測方法進行了更新,通過提高檢測方法的敏感性確保產(chǎn)品中殘留DNA的安全性。中國藥典中的檢測方法與美國、歐洲各不相同[15-17]。因此,中國相關器械企業(yè)在研發(fā)與生產(chǎn)中應具有前瞻性,盡早更新相關檢測方法,這將有利于改進工藝、提高產(chǎn)品質量,有利于產(chǎn)品走出國門提高產(chǎn)品的競爭力。
動物源性醫(yī)療器械的風險評估是基于風險-收益權重的產(chǎn)品安全性和有效性分析過程。盡管監(jiān)管機構和生產(chǎn)企業(yè)將殘留DNA作為進行產(chǎn)品質量控制的性能指標,但任何生物過程中殘留DNA的水平還是關鍵的安全性風險,為制造過程和產(chǎn)品上市提供安全性保證。對于動物源性醫(yī)療器械中殘留DNA帶來的風險隱患,采用安全可靠的工藝手段去除DNA是降低風險、保證產(chǎn)品質量的關鍵。建立合適的殘留DNA檢測方法有助于監(jiān)測工藝質量,確保動物源性醫(yī)療器械產(chǎn)品的安全性和有效性,造福人類健康。
猜你喜歡
中國設備工程(2022年12期)2022-07-11 04:33:00
中學生數(shù)理化·七年級數(shù)學人教版(2021年6期)2021-11-22 07:50:58
中學生數(shù)理化·七年級數(shù)學人教版(2021年6期)2021-11-22 07:50:58
中學生數(shù)理化·七年級數(shù)學人教版(2021年6期)2021-11-22 07:50:58
中學生數(shù)理化·七年級數(shù)學人教版(2020年12期)2021-01-18 06:57:46
中學生數(shù)理化·七年級數(shù)學人教版(2020年12期)2021-01-18 06:57:46
中學生數(shù)理化·七年級數(shù)學人教版(2019年9期)2019-11-25 07:34:36
中學生數(shù)理化·七年級數(shù)學人教版(2019年9期)2019-11-25 07:34:34
中學生數(shù)理化·七年級數(shù)學人教版(2019年12期)2019-05-21 02:53:50
中學生數(shù)理化·七年級數(shù)學人教版(2019年12期)2019-05-21 02:53:48
