拉米夫定替諾福韋片有關物質HPLC方法檢測
2023-12-11 10:29:47唐嘉悅黃德見
赤峰學院學報·自然科學版 2023年11期
關鍵詞:質量控制
唐嘉悅 黃德見
摘 要:建立HPLC法檢測拉米夫定替諾福韋片有關物質的方法,為拉米夫定替諾福韋片的質量控制提供方法依據。采用BDS C18色譜柱(250mm×4.6mm,5μm),以0.025mol/L醋酸銨、乙腈和甲醇為流動相,梯度洗脫,流速1.0mL/min,柱溫35℃,檢測波長260nm,進樣量5μL。結果顯示各色譜峰均能良好分離,各雜質濃度和峰面積線性關系良好,平均回收率在95.0%~105.0%。本方法專屬性強、靈敏、準確,可用于拉米夫定替諾福韋片有關物質的測定,同時填補了國內關于拉米夫定替諾福韋片有關物質檢測方法的空白。
關鍵詞:拉米夫定替諾福韋片;有關物質;質量控制
中圖分類號:R927.11? 文獻標識碼:A? 文章編號:1673-260X(2023)11-0016-04
拉米夫定替諾福韋片為拉米夫定和富馬酸替諾福韋二吡呋酯的復方制劑,臨床上主要用于乙型肝炎感染治療和艾滋病治療[1-4]。為抗人類免疫缺陷病毒(HIV)的一線藥物。WHO藥物資格預審名單中收錄有Hetero、Mylan、Cipla等六家印度公司生產本品,截至2019年國內僅1家公司被批準上市。
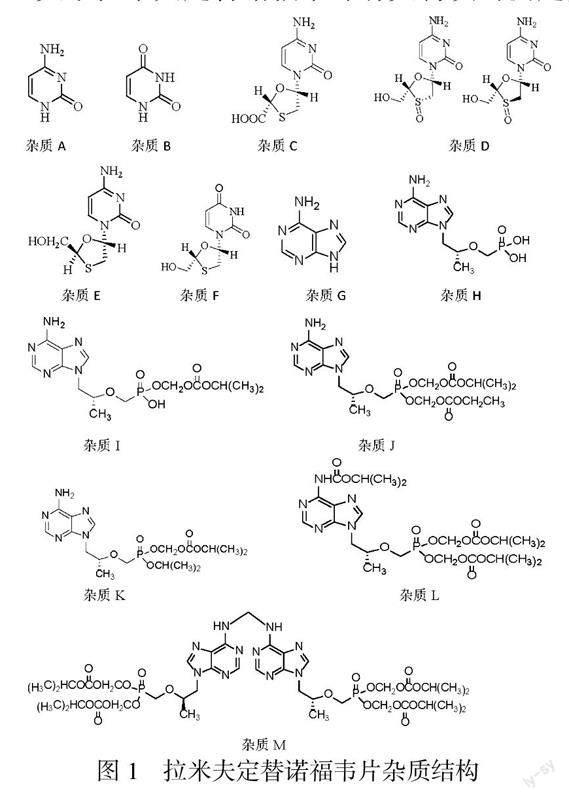
關于拉米夫定替諾福韋片有關物質的測定方法,國內外未見文獻報道。為了嚴格控制其質量,本文采用HPLC法對拉米夫定替諾福韋片有關物質進行了研究,結合拉米夫定和富馬酸替諾福韋二吡呋酯的合成工藝,分析其雜質譜[5-8],其結構式見圖1。
1 儀器與試藥
1.1 儀器
Agilent 1260型高效液相色譜儀,安捷倫科技(中國)有限公司產;BT25S電子天平,賽多利斯科學儀器(北京)有限公司產。
1.2 試藥及試劑
對照品:富馬酸替諾福韋二吡呋酯(GOM335,USP,99.3%)、拉米夫定(210405,自制,99.5%)、拉米夫定分離度混合物C(USP,FOK348)、雜質對照品A(210305,自制,98.5%)、雜質對照品B(210305,自制,99.5%)、雜質對照品C(210305,自制,97.5%)、雜質對照品D(210305,自制,98.4%)、雜質對照品E(210306,自制,98.5%)、雜質對照品F(210205,自制,99.2%)、雜質對照品G(210305,自制,95.5%)、雜質對照品H(210305,自制,96.7%)、雜質對照品I(210305,自制,98.5%)、雜質對照品J(210315,自制,98.5%)雜質對照品K(210325,自制,96.5%)雜質對照品L(210308,自制,97.5%)雜質對照品M(210205,自制,99.5%)。
試劑:乙腈(色譜純)、水(超純水)、醋酸銨(分析純)、冰醋酸(分析純)。
2 方法與結果
2.1 色譜條件
色譜柱:BDS C18柱,4.6×250mm,5μm;檢測波長260nm;柱溫35℃;流速1.0mL/min;進樣溫度6℃;進樣量5μL。
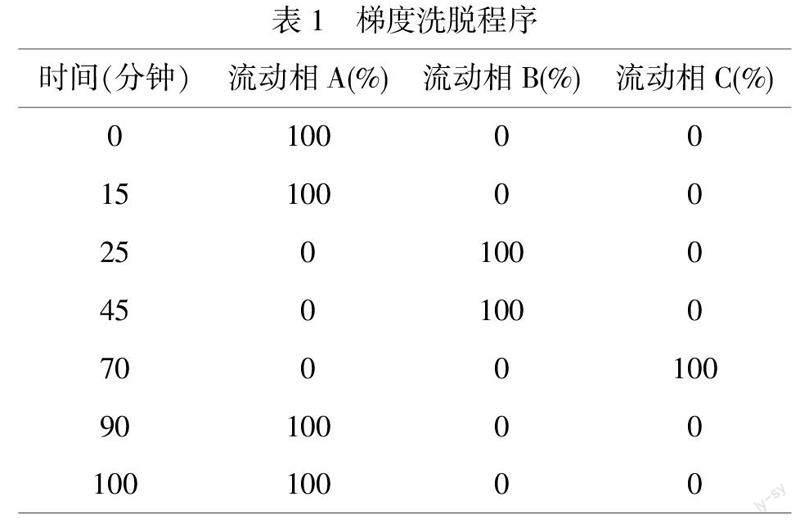
流動相:按表1進行線性梯度洗脫;
流動相A-0.025mol/L醋酸銨(用冰醋酸調節pH值至3.9±0.1):甲醇(95:5)
流動相B-0.025mol/L醋酸銨(用冰醋酸調節pH值至3.9±0.1):乙腈(40:60)
流動相C-0.025mol/L醋酸銨(用冰醋酸調節pH值至3.9±0.1):乙腈(20:80)
2.2 溶液配制
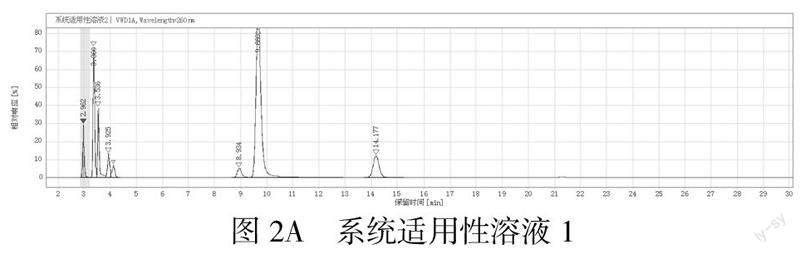
(1)系統適用性溶液的配制。取拉米夫定分離度混合物C對照品1支,加入乙腈:水(10:90)混合溶劑2.5mL使溶解,搖勻,作為系統適用性溶液(1)。
取拉米夫定,富馬酸替諾福韋二吡呋酯和雜質G、H、I、J、K、L、M各適量,加入流動相A溶解稀釋定容后配制成含拉米夫定3mg/mL、富馬酸替諾福韋二吡呋酯3mg/mL,雜質I 100μg/mL其他雜質10μg/mL的混合溶液,作為系統適用性溶液(2)。
(2)供試品溶液的配制。精密稱取本品細粉適量(約相對于富馬酸替諾福韋二吡呋酯30mg),置10mL量瓶中,加流動相A適量超聲5分鐘,并稀釋至刻度,搖勻,用0.45?滋m的濾膜濾過。
(3)對照溶液的配制。精密量取供試品溶液1mL,置200mL量瓶中,用流動相A稀釋至刻度,搖勻。
2.3 方法學驗證
2.3.1 系統適用性
取系統適用性溶液(1)(2)各5?滋L按上述色譜條件連續進樣5次,結果見圖2。結果顯示,各色譜峰之間分離度均大于1.5。重復進樣結果顯示各組分系統參數的RSD%均≤5.0%;結果均符合要求。
2.3.2 專屬性實驗
精密量取流動相A 5μL,按上述色譜條件注入液相色譜儀,按處方比例取混合輔料按供試品溶液制備方法制成溶液,照上述色譜條件測定,結果空白溶劑空白輔料均不干擾樣品檢測。結果見圖3。
(1)酸破壞試驗:精密稱取本品的細粉適量(約相當于富馬酸替諾福韋二吡呋酯30mg),置10mL量瓶中,加6mol/L鹽酸溶液2ml,搖勻,放置10分鐘,中和,加流動相A適量使溶解并稀釋至刻度,搖勻,用0.45μm的濾膜濾過。
(2)堿破壞試驗:精密稱取本品的細粉適量(約相當于富馬酸替諾福韋二吡呋酯30mg),置10mL量瓶中,加0.1mol/L氫氧化鈉溶液2mL,搖勻,放置10分鐘,中和,加流動相A適量使溶解并稀釋至刻度,搖勻,用0.45μm的濾膜濾過。
(3)高溫破壞試驗:精密稱取置100℃加熱6h樣品細粉適量(約相當于富馬酸替諾福韋二吡呋酯30mg),置10mL量瓶中,加流動相A適量使溶解并稀釋至刻度,搖勻,用0.45μm的濾膜濾過。
(4)氧化破壞試驗:精密稱取本品的細粉適量(約相當于富馬酸替諾福韋二吡呋酯30mg),置10mL量瓶中,加30%過氧化氫溶液2mL,搖勻,放置10分鐘,加流動相A適量使溶解并稀釋至刻度,搖勻,用0.45μm的濾膜濾過。
(5)光照破壞試驗:精密稱取已置太陽光下照射15天的本品細粉適量(約相當于富馬酸替諾福韋二吡呋酯30mg),置10mL量瓶中,加流動相A適量使溶解并稀釋至刻度,搖勻,用0.45μm的濾膜濾過。
按上述色譜條件進樣,記錄色譜圖。結果顯示,主峰與相鄰雜質及各降解雜質峰分離度符合要求,結果見表2。
2.4 檢測限、定量限
將上述雜質用流動相不斷稀釋,精密量取檢測限、定量限溶液各5μL按2.1項下色譜條件注入液相色譜儀,當信噪比S/N≥10時,即為定量濃度及定量限,當信噪比S/N≥3時,即為檢測濃度及檢測限。結果雜質A的定量限濃度為0.1679μg/mL,檢測限濃度為0.0569μg/mL;雜質B的定量限濃度為0.1532μg/mL,檢測限濃度為0.0528μg/mL;雜質C的定量限濃度為0.2000?g/mL,檢測限濃度為0.1000μg/mL;雜質D的定量限濃度為0.2000μg/mL,檢測限濃度為0.1000μg/mL;雜質E的定量限濃度為0.1500μg/mL,檢測限濃度為0.0785μg/mL;雜質F的定量限濃度為0.1500μg/mL,檢測限濃度為0.0750μg/mL;雜質G的定量限濃度為0.1500μg/mL,檢測限濃度為0.0700μg/mL;雜質H的定量限濃度為0.1500μg/mL,檢測限濃度為0.0785μg/mL;雜質I的定量限濃度為0.2500μg/mL,檢測限濃度為0.0569μg/mL;雜質J的定量限濃度為0.1500μg/mL,檢測限濃度為0.0785μg/mL;雜質L的定量限濃度為0.1500μg/mL,檢測限濃度為0.0785μg/mL;雜質M的定量限濃度為0.1500μg/mL,檢測限濃度為0.0785μg/mL;拉米夫定的定量限濃度為0.1750μg /mL,檢測限濃度為0.0890μg/mL;富馬酸替諾福韋二吡呋酯的定量限濃度為0.1750μg/mL,檢測限濃度為0.0875μg/mL。
2.5 線性
取上述雜質對照品各適量,精密稱定,加流動相A使溶解,再加稀釋劑逐步稀釋制成適宜濃度的系列標準溶液。精密量取系列標準溶液各5μL,按上述色譜條件注入液相色譜儀,記錄峰面積。以C(μg/mL)為橫坐標,以峰面積為縱坐標,做回歸方程。結果雜質A的回歸方程為A=9.7014C-0.314 r=1;雜質B的回歸方程為A=56.5977C+0.2359 r=0.9999;雜質C的回歸方程為A=56.5977C+0.2359 r=0.9999;雜質D的回歸方程為A=8.9358C-0.7073 r=1;雜質E的回歸方程為A=7.1591C-1.602 r=1;雜質F的回歸方程為A=14.029C+1.0604 r=0.9998;拉米夫定的回歸方程為A=9.7727C+8.7101 r=0.9999;富馬酸替諾福韋二吡呋酯的回歸方程為A=6.0942C+39.024 r=0.9998;雜質G的回歸方程為A=6.2244C+8.1014 r=1;雜質H的回歸方程為A=28.288C-0.7321 r=0.9998;雜質I的回歸方程為A=16.953C-2.2805 r=1;雜質J的回歸方程為A=10.352C+1.42263 r=0.9999;雜質K的回歸方程為A=8.7401C-2.6152 r=1;雜質L的回歸方程為A=14.703C-1.165 r=0.9997;雜質M的回歸方程為A=20.138C-1.2967 r=1,各組分在LOQ~至少150%的限量濃度范圍內,峰面積與濃度呈良好的線性關系。
2.6 重復性
取同一批號樣品適量,共6份,按供試品溶液的制備方法制成供試品溶液,按上述色譜條件注入液相色譜儀,結果6次結果顯示雜質檢出個數、已知雜質檢出量、最大單雜和總雜質基本不變;說明本方法具有較好的重復性。
2.7 回收率
取同一批號樣品適量,分別置于9個10mL量瓶中,再分別按限度濃度的80%、100%和120%加入上述對照品各3份混勻,按上述供試品溶液的制備方法制成供試品溶液。結果,雜質A、雜質B、雜質C、雜質D、雜質E、雜質F、雜質G、雜質H、雜質I、雜質J、雜質K、雜質L、雜質M的回收率分別為96.5%、103.9%、100.1%、103.6%、102.9%、103.5%、98.5%、99.5%、97.2%、96.5%、98.2%、94.8%、96.5%。RSD%分別為0.30%、0.45%、1.85%、0.42%、1.58%、2.15%、1.24%、1.38%、1.45%、2.10%、1.98%、2.00%、1.87%。
2.8 耐用性
分別考察了醋酸銨的濃度、pH值、流速、柱溫以及流動相比例的±10%微小變化對系統適用性(1)(2)及同一批次有關物質供試品溶液檢測結果的影響,結果各因素微小變化對系統適用性試驗及有關物質檢測結果基本無影響。說明本方法耐用性良好。
3 討論
3.1 檢測波長的選擇
富馬酸替諾福韋二吡呋酯和拉米夫定最大吸收波長分別為260nm和277nm,由于兩者處方量相同,綜合各雜質的紫外吸收,對波長260nm和277nm分別進行了專屬性考查,結果表明通過酸、堿、氧化、高溫、光照強制降解試驗主峰與相鄰雜質峰之間均能較好分離。考慮到在相同濃度下富馬酸替諾福韋二吡呋酯在277nm波長處吸收比拉米夫定弱。因此選擇富馬酸替諾福韋二吡呋酯最大吸收波長260nm作為檢測波長[9-12]。
3.2 流動相及色譜柱的選擇
本品為復方制劑,建立有關物質分析方法時應同時考慮拉米夫定和富馬酸替諾福韋二吡呋酯相關雜質,由于拉米夫定及其雜質極性大于富馬酸替諾福韋二吡呋酯及相關雜質,因此本方法前15分鐘主要參考“拉米夫定片”中國藥典或BP藥典方法用于對拉米夫定及相關雜質的分離。至15分鐘后,隨著有機相比例增高,富馬酸替諾福韋二吡呋酯相關雜質可被完全洗脫出來。最終優化方法色譜條件下,拉米夫定的保留時間約為9分鐘,富馬酸替諾福韋二吡呋酯的保留時間約為38分鐘。各峰之間分離度均較好[13-15]。
4 結論
本實驗建立了一種有效檢測拉米夫定替諾福韋片有關物質的分析方法,并對其進行了方法學驗證。實驗結果表明,該方法可簡便有效地檢測相關雜質,各雜質之間的分離度均大于1.5。系統適用性、專屬性、線性、回收率均較好,檢測靈敏度高。可用于拉米夫定替諾福韋片有關物質的檢測。填補了國內關于該品種有關物質檢測方法的空白,為質量標準制定奠定了基礎。
參考文獻:
〔1〕余史丹,汪碩聞,唐原君,等.替諾福韋、依非韋倫對拉米夫定在大鼠體內藥動學的影響[J].中南藥學,2022,20(09):2033-2040.
〔2〕馬春濤,霍秀敏,王強.我國艾滋病抗病毒治療藥品注冊情況分析[J].中國艾滋性病,2020,26(09):941-944+953.
〔3〕項少黎,儲文龍.替諾福韋聯合拉米夫定片和洛匹那韋利托那韋片治療艾滋病的效果評價[J].中國實用醫藥,2022,17(05):163-165.
〔4〕黃婉盈,郭姝悅,張曉丹.替諾福韋酯聯合拉米夫定治療艾滋病合并乙型肝炎病毒感染的臨床療效分[J].航空航天醫學雜志,2021,32(09):1094-1095.
〔5〕徐鳳杰,楊永忠.拉米夫定的合成進展[J].云南化工,2009,36(06):33-35.
〔6〕王華,鄒柳,劉映,等.拉米夫定的合成工藝優化[J].臨床合理用藥雜志,2016,9(34):88-90.
〔7〕朱衛兵.富馬酸替諾福韋二吡呋酯的合成[J].應用化工,2020,49(S2):360-362.
〔8〕張奇,郭明,赫暢,等.富馬酸替諾福韋二吡呋酯的合成工藝[J].沈陽藥科大學學報,2018,35(07):543-546+556.
〔9〕伏瑤,劉松霞,范彩霞,等.伊沙匹隆有關物質的HPLC法檢測[J].藥物分析雜志,2022,42(10):1801-1807.
〔10〕劉曉坤,王世瀟,方詩源,等.高效液相色譜法同時測定普那布林原料藥中9種有關物質[J].中國海洋藥物,2023,42(03):63-68.
〔11〕劉振麗,徐麗.高效液相色譜法測定西咪替丁片中的有關物質和含量[J].黑龍江醫藥,2022,46(15):1854-1856.
〔12〕白莉霞,劉希望,楊亞軍,等.高效液相色譜法測定阿司匹林丁香酚酯咀嚼片的有關物質[J].中國畜牧醫藥,2023,50(07):3007-3016.
〔13〕龐賽,梁愛仙,陳顏清,等.HPLC法測定氨酚曲馬多片有關物質方法的建立[J].中國藥師,2022, 25(09):1689-1693.
〔14〕周琳.煙酸片中有關物質的測定及結果分析[J].海峽藥學,2022,34(05):71-74.
〔15〕保敏敏,呂蓓蓓,魏文芝,等.奧卡西平片中有關物質含量測定方法的建立[J].中國藥房,2023,34(10):1199-1203.
猜你喜歡
中國科技博覽(2016年19期)2016-10-19 13:36:59
中國科技博覽(2016年18期)2016-10-19 11:06:33
中國科技博覽(2016年18期)2016-10-19 09:03:36
中國科技博覽(2016年18期)2016-10-19 08:46:18
科技視界(2016年21期)2016-10-17 17:58:28
中國實用醫藥(2016年24期)2016-10-17 06:28:30
科學與財富(2016年28期)2016-10-14 19:44:52
科學與財富(2016年28期)2016-10-14 18:58:41
科學與財富(2016年28期)2016-10-14 18:44:58
科技視界(2016年20期)2016-09-29 13:11:33
