綠原酸對牙齦卟啉單胞菌誘導牙齦上皮細胞自噬的調控作用及機制*
2023-12-23 05:26:28吳珊趙國廷董振耀馬秀花姚毅章
西部醫學 2023年12期
吳珊 趙國廷 董振耀 馬秀花 姚毅章
(青海省第五人民醫院口腔科,青海 西寧810007)
牙齦卟啉單胞菌(Porphyromonas gingivalis,P.g)是牙周炎的致病菌,也是口腔鱗狀細胞癌的危險因素。研究顯示,P.g感染可通過破壞牙齦上皮屏障、內化進入牙齦上皮細胞,促進牙齦上皮細胞增殖和自噬[1-2]。自噬是一種細胞內降解過程,在維持細胞穩態方面起著關鍵作用,據報道,P.g可誘導牙齦上皮細胞自噬,利用自噬機制來生存[3];且自噬可誘導破骨細胞增殖,導致牙槽骨吸收,這是導致牙周炎患者牙齒脫落的根本原因[4]。因此,抑制P.g誘導的自噬是治療牙周病的一個新策略。AMP活化蛋白激酶(AMP-activated protein kinase,AMPK)/UNC-51樣激酶1(UNC-51-like kinase 1,ULK1)通路在調節細胞自噬過程中發揮重要作用,激活的AMPK可使ULK1磷酸化,誘導自噬,而抑制AMPK/ULK1通路激活,可抑制自噬體的產生[5]。綠原酸(Chlorogenic acid,CGA)是具有多種藥理活性的酚類化合物,可從金銀花、杜仲等藥材獲得,具有抗炎、抗氧化作用。研究顯示,負載CGA的納米膠束在局部注射后可在牙齦組織中保留超過24 h,抑制牙槽骨吸收并阻止牙周炎小鼠模型進展,有望成為牙周炎的新型治療策略[6]。此外,CGA被報道可抑制P.g的增殖和P.g的蛋白酶活性,對牙周疾病發揮保護作用[7]。然而,CGA對P.g誘導的牙齦上皮細胞自噬的影響及作用機制尚不明確。研究顯示,CGA能夠通過激活AMPK發揮降糖調脂的抗糖尿病作用[8];CGA還可抑制皮質酮誘導的PC12細胞的自噬與凋亡,發揮抗抑郁作用[9]。以上研究表明CGA對AMPK通路和自噬具有調節作用,但CGA是否通過調節AMPK/ULK1通路影響人牙齦上皮細胞的自噬與凋亡少見報道。因此,本研究通過P.g誘導人牙齦上皮細胞(Human gingival epithelial cells,HGE)建立炎性HGE細胞模型,探討CGA對炎性HGE細胞自噬的影響及其可能的作用機制,為牙周病的治療提供理論基礎。
1 材料與方法
1.1 實驗材料與主要試劑 HGE、牙齦卟啉單胞菌(P. gingivalis)(ATCC,美國),CGA(HPLC≥98%,源葉生物科技有限公司),MTT檢測試劑盒、Annexin V-FITC/PI細胞凋亡檢測試劑盒、BCA蛋白檢測試劑盒(北京百奧萊博科技有限公司),AICAR(Selleck,美國),兔抗人LC3Ⅱ、LC3I、Beclin-1、P62、p-ULK1、ULK1、p-AMPK、AMPK抗體及Alexa Fluor?488標記的二抗(Abcam,英國)。
1.2 細胞增殖實驗 HGE細胞培養于含藥雙抗(100 U/mL青霉素、100 μg/mL鏈霉素)的RPMI1640培養液中,待細胞生長至對數期時,以每孔2×104個細胞接種于96孔板中,使用或不使用P.g處理(MOI=100)HGE細胞12 h,再使用濃度為5、10、20、40、60 mg/L的CGA處理24 h,每孔加入20 μL的MTT溶液(PBS緩沖液制成5 mg/mL溶液),37 ℃避光孵育4 h,再加入150 μL DMSO溶液,檢測酶標儀490 nm處吸光度值。
1.3 細胞處理及分組 將對數生長期HGE細胞分為對照組(Control組,不做任何處理)、P.g組、CGA組(藥物濃度分別為10.0、20.0、40.0 mg/L)、CGA+AICAR(AMPK激活劑)組,每組設置6個復孔。P.g組使用P.g誘導建立細胞模型;CGA組在P.g誘導HGE細胞12 h后,分別使用終濃度為10.0、20.0、40.0 mg/L CGA(溶于DMSO中配制成100 mg/mL母液)處理細胞24 h;CGA+AICAR組P.g誘導HGE細胞12 h后,使用濃度為40.0 mg/L的CGA與1.0 nmol/L的AICAR共同處理細胞24 h,Control組與P.g組不使用藥物處理。
1.4 流式細胞儀檢測細胞凋亡 各組處理的HGE細胞以每孔2×104個細胞接種于96孔板中培養24 h后,0.25%胰蛋白酶消化細胞,PBS洗滌,加入5 μL FITC染料及2.5 μL PI染料,避光孵育20 min,流式細胞儀檢測細胞凋亡。
1.5 免疫熒光染色檢測HGE細胞LC3Ⅱ表達 各組處理的HGE細胞使用冰PBS緩沖液洗滌細胞,4%預冷的多聚甲醛固定10 min,冰PBS洗滌3次,0.5%的TritonX-100孵育細胞10 min,冰PBS洗滌3次,5% BSA室溫封閉30 min,加入LC3Ⅱ抗體(5%BSA配制)4 ℃孵育過夜,加入Alexa Fluor?488標記的二抗室溫避光孵育1h,DAPI復染細胞核,熒光顯微鏡觀察并拍照。
1.6 Western blot檢測蛋白表達 各組處理的HGE細胞以每孔1×106接種于6孔板培養48 h后,500×g離心5 min、收集細胞,加入預冷的RIPA裂解液提取蛋白,使用BCA試劑盒檢測蛋白含量,取30 μg蛋白進行電泳分離并轉膜,5%的BSA封閉1 h,別加入LC3Ⅱ(1:1000)、LC3I(1:1000)、Beclin-1(1:1000)、P62(1:1500)、p-ULK1(1:1500)、ULK1(1:1000)、p-AMPK(1:1000)、AMPK(1:1000)稀釋液和β-actin(1:1500)抗體稀釋液,4 ℃孵育過夜,加入二抗室溫孵育1h,ECL顯色放大,凝膠成像系統成像,以β-actin為內參,對各蛋白進行定量分析。
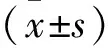
2 結果
2.1 CGA對HGE細胞增殖的影響 與Control組比較,使用5、10、20、40、60 mg/L的CGA處理HGE細胞24 h后,細胞增殖活性無顯著變化(P>0.05),見圖1。
2.2 CGA對炎性HGE細胞增殖的影響 與Control組比較,P.g處理HGE細胞后,細胞增殖活性顯著增加,使用5~60 mg/L的CGA處理P.g誘導的HGE細胞24 h后,細胞增殖活性有所下降,CGA濃度在10 mg/L以上時,對HGE細胞的抑制作用較為明顯(圖2),故選擇10、20、40 mg/L的CGA進行后續實驗。
2.3 CGA對炎性HGE細胞凋亡的影響 與Control組比較,P.g組細胞凋亡率差異無統計學意義(P>0.05);與P.g組比較,濃度為10.0、20.0、40.0 mg/L的CGA處理的HGE細胞凋亡率顯著升高,且呈藥物濃度依賴性(P<0.05);與40mg/L CGA組比較,CGA+AICAR組HGE細胞凋亡率顯著降低(P<0.05)。見圖3。

圖3 CGA對炎性HGE細胞凋亡的影響
2.4 CGA對炎性HGE細胞LC3Ⅱ表達的影響 免疫熒光染色結果顯示,LC3Ⅱ顯示為綠色熒光,與Control組比較,P.g組綠色熒光增強,相對熒光強度顯著升高(P<0.05);與P.g組比較,CGA濃度為10.0、20.0、40.0 mg/L處理組HGE細胞綠色熒光明顯減弱,相對熒光強度顯著降低(P<0.05);與40mg/L CGA組比較,CGA+AICAR組綠色熒光增強,相對熒光強度顯著升高(P<0.05),見圖4。

圖4 免疫熒光檢測炎性HGE細胞LC3Ⅱ表達
2.5 CGA對炎性HGE細胞自噬蛋白表達的影響 與Control組比較,P.g組LC3Ⅱ/I、Beclin-1蛋白表達水平顯著升高,P62表達水平顯著降低(P<0.05);與P.g組比較,濃度為10.0、20.0、40.0 mg/L的CGA處理組LC3Ⅱ/I、Beclin-1蛋白表達水平顯著降低,P62表達水平顯著升高(P<0.05),且呈藥物濃度依賴性;與40.0 mg/L CGA組比較,CGA+AICAR組HGE細胞LC3Ⅱ/I、Beclin-1蛋白表達水平顯著升高,P62表達水平顯著降低(P<0.05),見圖5。

圖5 Western blot檢測炎性HGE細胞LC3Ⅱ/I、Beclin-1、P62蛋白表達
2.6 CGA對炎性HGE細胞AMPK/ULK1通路相關蛋白表達的影響 與Control組比較,P.g組p-ULK1、p-AMPK蛋白表達水平顯著升高(P<0.05);與P.g組比較,濃度為10.0、20.0、40.0 mg/L的CGA處理組p-ULK1、p-AMPK蛋白表達水平顯著降低(P<0.05),且呈藥物濃度依賴性;與40.0 mg/L CGA組比較,CGA+AICAR組HGE細胞p-ULK1、p-AMPK蛋白表達水平顯著升高(P<0.05),見圖6。

圖6 Western blot檢測炎性HGE細胞p-ULK1、p-AMPK蛋白表達
3 討論
P.g可誘導口腔來源細胞如牙齦上皮細胞、牙齦成纖維細胞的自噬,實現其在宿主細胞內的生存和增殖,進而促進牙周炎的炎性反應。脂多糖是P.g外膜的重要毒力因子,可激活細胞炎癥反應與自噬[10]。本研究使用P.g誘導HGE細胞后,細胞自噬水平升高,表明建模成功。
CGA是一種由咖啡酸和奎尼酸組成的縮酚酸化合物,具有抗炎、抗氧化、抗腫瘤、抗病毒的作用,還具有調節糖脂代謝,治療代謝疾病的作用[11-12]。研究顯示,CGA可抑制高糖誘導的人牙周膜成纖維細胞凋亡[13];且CGA可抑制P.g的增殖,表現出抗菌作用,還可抑制P.g的蛋白酶活性,對牙周疾病具有保護作用[7]。本研究使用5~60 mg/L的CGA處理炎性HGE細胞24 h后,HGE細胞增殖活性下降,表明CGA對炎性HGE細胞增殖有抑制作用,且CGA濃度在10 mg/L及其以上時,可顯著抑制HGE細胞的增殖,故選擇濃度為10、20、40 mg/L的CGA進行后續實驗。
CGA可調節細胞自噬,自噬與細胞凋亡密切相關,如CGA可通過上調溶酶體功能增強自噬保護過氧化氫誘導的人神經母細胞瘤細胞氧化損傷[14];而在皮質酮誘導的PC12細胞中,CGA可通過抑制自噬,抑制PC12細胞凋亡[9]。本研究使用10、20、40 mg/L的CGA處理炎性HGE細胞,發現CGA可呈濃度依賴性方式誘導HGE細胞凋亡。LC3Ⅱ、Beclin-1、P62自噬的標志性蛋白,P.g被報道可促進人牙齦成纖維細胞自噬,誘導LC3Ⅱ表達[15]。本研究使用不同濃度的CGA處理炎性HGE細胞后,LC3Ⅱ、Beclin-1蛋白表達水平顯著降低,P62表達水平顯著升高,且呈濃度依賴性,表明CGA可抑制炎性HGE細胞自噬,誘導細胞凋亡。這與既往研究中CGA誘導自噬的結果不一致,推測其原因可能與自噬的特性有關。因為自噬是一把雙刃劍,即可誘導凋亡,又可抑制凋亡,這種抑制或促進作用可以根據細胞內情形而相互轉化。
細胞自噬由多種信號通路調控,AMPK是細胞能量感受器,也是應激反應觸發自噬的主要調節因子[16-17];在能量缺乏下,AMPK發生磷酸化而被激活,活化的AMPK可以使自噬起始關鍵蛋白ULK1磷酸化,ULK1活化后,可提高FUNDC1與LC3Ⅱ的結合效率,促進自噬的產生[18]。AMPK/ULK1信號通路在多種疾病(如藥物神經損傷、腦缺血、肝損傷)的自噬中發揮重要調控作用[19-20]。研究顯示,抑制AMPK/ULK1通路激活可抑制缺氧復氧誘導的星形膠質細胞自噬,促進細胞存活并減少細胞凋亡[21]。而激活AMPK/ULK1通路,可誘導心肌細胞自噬與凋亡[22]。本研究使用不同濃度CGA處理炎性HGE細胞后,p-ULK1、p-AMPK蛋白水平降低,且呈濃度依賴性,提示CGA具有抑制AMPK/ULK1通路激活的作用。此外,本研究還顯示,使用AMPK激活劑AICAR與CGA共同處理炎性HGE細胞后,HGE細胞凋亡率下降,LC3Ⅱ、Beclin-1蛋白水平顯升高,P62表達水平降低,表明AICAR可部分逆轉CGA對炎性HGE細胞自噬的抑制作用。以上結果表明,CGA可能通過抑制AMPK/ULK1通路,抑制人牙齦上皮細胞自噬,促進細胞凋亡。但姚宏紀等[8]的報道顯示,CGA對AMPK發揮激動作用,在糖尿病合并糖脂代謝紊亂的小鼠中,CGA可通過激活AMPK改善血脂、糖耐量和胰島素抵抗。這與本研究結果不一致,推測CGA對AMPK是發揮激動還是抑制作用與疾病、細胞和組織類型有關。
4 結論
CGA可通過抑制AMPK/ULK1通路抑制炎性HGE細胞自噬與增殖,并促進細胞凋亡。本研究初步探討了CGA作為牙周炎治療候選藥物的潛力,但CGA的具體作用與機制尚需進一步的研究驗證,在未來的研究中將以牙齦成纖維細胞為對象并結合體內實驗,進行深入分析。此外,CGA在機體的代謝與安全性評估也是接下來的研究重點。
猜你喜歡
體育科技文獻通報(2022年3期)2022-05-23 13:46:54
中學生數理化·七年級數學人教版(2021年6期)2021-11-22 07:50:58
中學生數理化·七年級數學人教版(2021年6期)2021-11-22 07:50:58
中學生數理化·七年級數學人教版(2021年6期)2021-11-22 07:50:58
天津外國語大學學報(2021年3期)2021-08-13 08:32:18
遼金歷史與考古(2021年0期)2021-07-29 01:06:54
中學生數理化·七年級數學人教版(2020年12期)2021-01-18 06:57:46
中學生數理化·七年級數學人教版(2020年12期)2021-01-18 06:57:46
科技傳播(2019年22期)2020-01-14 03:06:54
民用飛機設計與研究(2019年4期)2019-05-21 07:21:24
