液相色譜串聯質譜法同時測定凡納濱對蝦中的27種抗寄生蟲藥物殘留
2023-12-28 01:01:10張麗芳馬瑞欣鄭百芹
食品與機械 2023年11期
張麗芳 馬瑞欣 鄭百芹 宋 旺 周 鑫
(1. 唐山市食品藥品綜合檢驗檢測中心,河北 唐山 063000;2. 農業農村部農產品品質評價與營養健康重點實驗室,河北 唐山 063000;3. 河北省水產技術推廣總站,河北 石家莊 050000)
抗寄生蟲藥物屬于獸藥,種類眾多,且作用機制各不相同,往往根據感染寄生蟲的類別或動物種類用藥[1-3],但不科學不規范的藥物施用方式會帶來一些負面作用,如土壤、水文等環境中藥物殘留的積累和遷移[4-5],寄生蟲、微生物等生物體產生耐藥性[6-7],藥物殘留隨食物鏈由食用性動物產品轉移到人體[8]等。為此,一些國際組織和國家紛紛制定了食品中獸藥殘留限量,并不斷更新和增補相關內容。中國制定的GB 31650—2019《食品安全國家標準 食品中獸藥最大殘留限量》標準規定了267種(類)獸藥在畜禽產品、水產品、蜂產品中的2 191項殘留限量及使用要求,依據藥物的安全性及其對食品安全的影響程度將獸藥(包括部分化學物質)進行了分類,但仍有一些常見的抗寄生蟲藥物種類不在標準名錄之中,需要進一步完善和修訂。
目前在抗寄生蟲藥物殘留研究中,較多集中在畜禽等陸地動物源性食品中,且藥物種類比較單一,而水產品中抗寄生蟲藥物殘留研究的報道相對較少。研究人員對水產品和環境中的獸藥[9-11]、農藥(包括抗菌劑)[12]等藥物做過相應的監測和評估,而以不同類型的抗寄生蟲藥物作為藥物殘留研究對象的報道并不多見,但是抗寄生蟲藥物作為畜禽養殖常用藥物通過環境遷移到水產養殖系統中并在水產動物中富集應予以重視。
由于凡納濱對蝦養殖場很多為非工廠化封閉養殖,養殖過程中可能通過排污、降水等形式受到附近周圍畜牧養殖場用藥污染和其他形式的藥物遷移,出于風險監控和排查的考慮需對凡納濱對蝦進行抗寄生蟲藥物殘留的測定。研究擬選取27種抗寄生蟲藥物作為凡納濱對蝦的藥物監測對象,使用QuEChERS對樣品進行提取和凈化,超高效液相串聯質譜定性定量分析,建立實驗室對凡納濱對蝦中抗寄生蟲藥物殘留測定方法,并對市場上銷售的凡納濱對蝦進行測定。
1 材料與方法
1.1 試驗試劑與耗材
甲醇、乙腈、甲酸:色譜純,美國Thermo Fisher公司;
甲酸銨:色譜純,美國Sigma-Aldrich公司;
氯化鈉:分析純,福晨(天津)化學試劑有限公司;
C18粉、乙二胺-N-丙基硅烷(primary secondary amine,PSA)粉、中性氧化鋁粉(Al2O3):北京迪馬科技有限公司;
0.22 μm濾膜:北京迪馬科技有限公司;
抗寄生蟲藥物液態標準品溶液(化合物名稱見表1):天津阿爾塔科技有限公司;
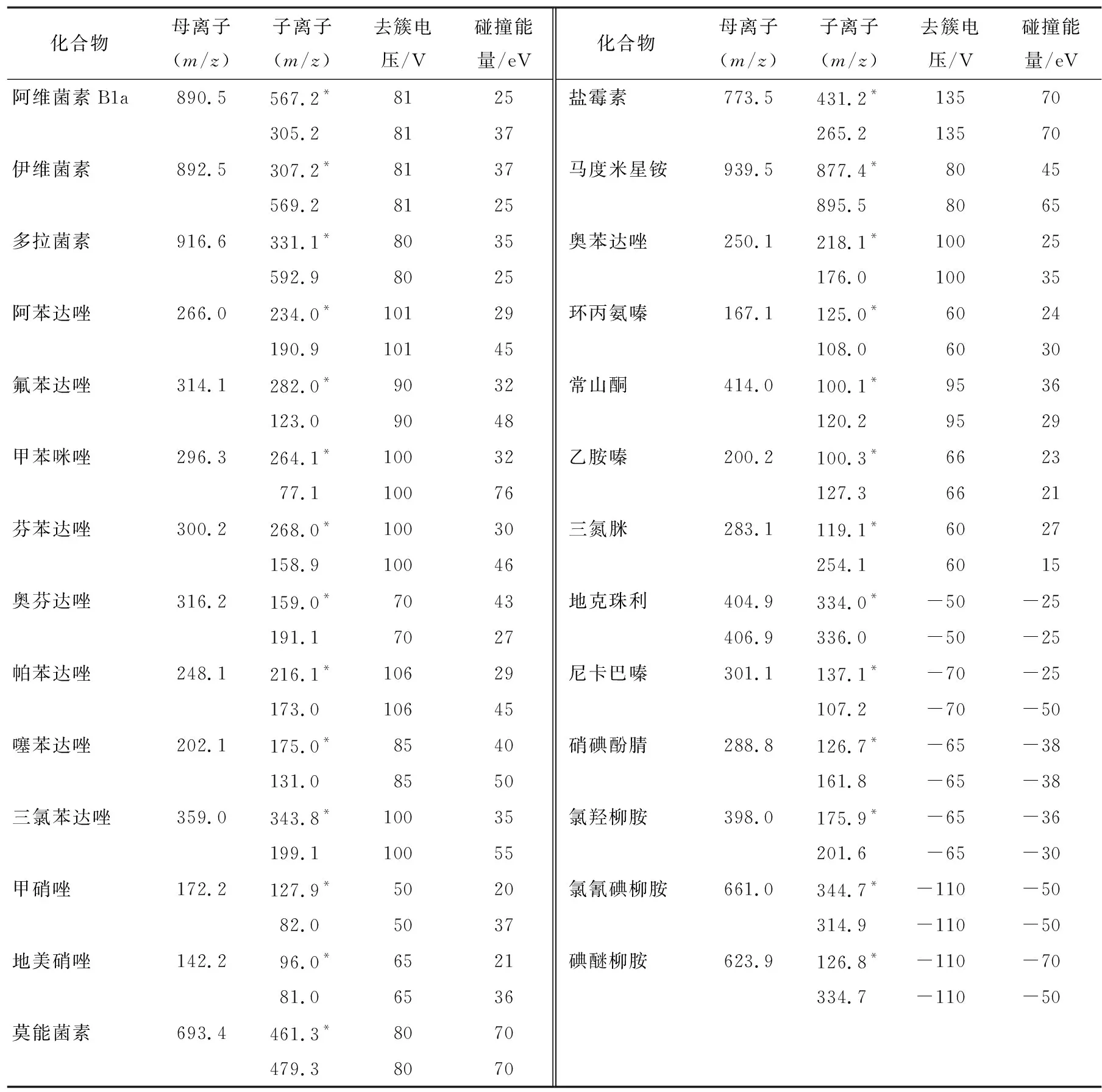
表1 化合物離子對信息及其對應質譜參數?
試驗用水為超純水。
1.2 儀器與設備
液相色譜串聯質譜儀:Triple Quad 5500+型 (配ESI離子源),美國AB SCIEX公司;
離心機:SORVALL LYNX 4000型,美國Thermo Fisher公司;
電子天平:BT 25 S型,德國Sartorius公司;
氮吹儀:EFAA-DC24-RT型,安譜實驗科技股份有限公司;
多位渦旋儀:KN-026S型,北京科德諾思技術有限公司。
1.3 標準溶液配制
吸取適量體積的27種抗寄生蟲藥物標準品溶液于容量瓶中,甲醇稀釋并定容,得到混合標準溶液,-20 ℃保存。
1.4 樣品前處理
稱取勻漿處理的蝦肉樣品2 g(精確至0.01 g),先后加入1 g氯化鈉和10 mL 80%乙腈水溶液,充分渦旋混合,超聲波提取10 min,于4 ℃下6 000 r/min離心5 min,準確吸取上層有機清液2 mL,加入C18和中性Al2O3粉末各100 mg,渦旋1 min充分吸附,取上清液過0.22 μm濾膜,待上機測定。
1.5 基質效應
配制溶劑稀釋標準曲線系列梯度和空白基質提取液稀釋標準曲線系列梯度,分別進樣上機測定并進行線性回歸分析,得到溶劑標準曲線和基質匹配標準曲線,按式(1)計算基質效應(matrix effect,ME)。
(1)
式中:
ME——基質效應數值;
km——基質匹配標準曲線斜率;
k——溶劑標準曲線斜率。
1.6 儀器分析條件
1.6.1 液相色譜條件 色譜柱:Thermo Hypersil Gold aQ (100×2.1 mm, 1.9 μm);柱溫:40 ℃;進樣量:2 μL;流速:0.4 mL/min。流動相:A,10 mmol/L甲酸銨(含0.1%甲酸)溶液;B,0.1%甲酸—乙腈。采用梯度洗脫:0.0~1.0 min,95% A,1.0~5.0 min,95%~5% A,5.0~8.0 min,5% A,8.0~8.1 min,5%~95% A,8.1~11.0 min,95% A。
1.6.2 質譜條件 離子源:電噴霧離子源(ESI);掃描模式:正負離子同步掃描模式;監測方式:多反應監測(MRM);噴霧電壓(IS):正掃描5 000 V,負掃描-4 000 V;離子源溫度550 ℃;氣簾氣(CUR):172 368.9 Pa;霧化氣(GS1):344 737.9 Pa;輔助氣(GS2):344 737.9 Pa;碰撞氣:7模擬單位。化合物離子對信息和相應其他質譜信息見表1。
2 結果與討論
2.1 質譜條件優化

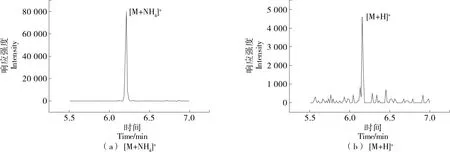
圖1 不同母離子選擇下多拉菌素的多反應監測響應強度比較
2.2 液相條件優化
2.2.1 色譜柱的選擇 選取Waters BEH C18(100 mm×2.1 mm,1.7 μm)、Thermo Hypersil Gold aQ(100 mm×2.1 mm,1.9 μm)和phenomenex Kinetex F5(100 mm×2.1 mm,2.6 μm)3種長度相同的色譜柱,在相同的條件下對目標化合物的分離情況進行考察。由圖2可知,Thermo Hypersil Gold aQ在物質分離程度和峰高的表現優于其他兩種,與劉開等[15]在色譜柱上的選擇不同,可能是由于化合物種類和數量以及所用的洗脫流動相不同所導致。研究選取Thermo Hypersil Gold aQ色譜柱作為分離用色譜柱,并進行后續試驗。

圖2 化合物在不同色譜柱上的分離情況
2.2.2 流動相的選擇 選取甲醇、乙腈、0.1%甲酸—乙腈作為有機相分別與作為水相流動相的水、0.1%甲酸水溶液、2 mmol/L甲酸銨溶液、2 mmol/L 甲酸銨溶液(含0.1%甲酸)、10 mmol/L甲酸銨溶液、10 mmol/L甲酸銨溶液(含0.1%甲酸)進行組合比較。作為有機相的甲醇和乙腈,乙腈更能夠將極性較弱的化合物從色譜柱中洗脫下來,而酸化流動相溶液可以促進咪唑類等藥物的離子化程度[16],與此同時,流動相中加入甲酸并未抑制負掃描化合物的離子化,響應信號未見明顯的降低。待測化合物中包含有阿維菌素類藥物,在母離子選擇上選取[M+NH4]+的母離子形式,流動相中加入銨鹽可以增強該類物質的離子化水平,并且可以改善色譜峰型。流動相中適宜的酸度和離子強度對于提高檢測靈敏度能起到積極作用[14],研究結合多種化合物性質和實際洗脫效果,最終選擇0.1%甲酸—乙腈和10 mmol/L 甲酸銨溶液(含0.1%甲酸)分別作為有機相和水相,采用梯度洗脫的形式對化合物進行分離。
2.3 前處理條件優化
2.3.1 提取試劑優化 選取甲醇、乙腈、80%乙腈水溶液、0.2%甲酸—乙腈對蝦肉中的抗寄生蟲藥物進行提取。通過試驗回收率對4種提取試劑進行考察,結果見圖3。由圖3可知,以甲醇作為抗寄生蟲藥物殘留的提取試劑時,雖然其中位值回收率最接近100%,但是各種藥物的提取效果呈現出分散的態勢,而且回收率大于120%或低于60%的藥物較多;以0.2%甲酸—乙腈作為提取試劑,藥物的回收率均低于100%,整體回收率過低,并且有9種藥物的回收率低于60%;以80%乙腈水溶液作為提取試劑時,各藥物回收率比較集中,主要分布在80%~100%。因此,選取80%乙腈水溶液作為提取試劑。
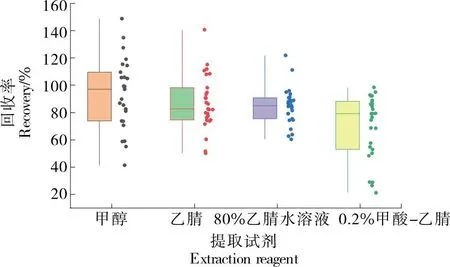
圖3 提取試劑對回收率的影響
2.3.2 凈化條件優化 C18、GCB(graphitic carbon)、PSA、Al2O3粉等是最常用的吸附劑[4]。PSA可以利用活性基上的氨基去除基質樣品中的糖、脂肪酸、有機酸、脂類、極性色素和金屬離子,C18對樣品中的脂肪和非極性成分具有良好的吸附作用;氧化鋁可以吸附脂肪酸等物質。試驗比較了C18、PSA和Al2O3粉對提取液的凈化效果。通過比較不同的吸附劑、吸附劑用量以及吸附劑的配伍應用,可以得出C18100 mg+Al2O3100 mg聯合應用吸附雜質的效果最佳,結果見圖4。
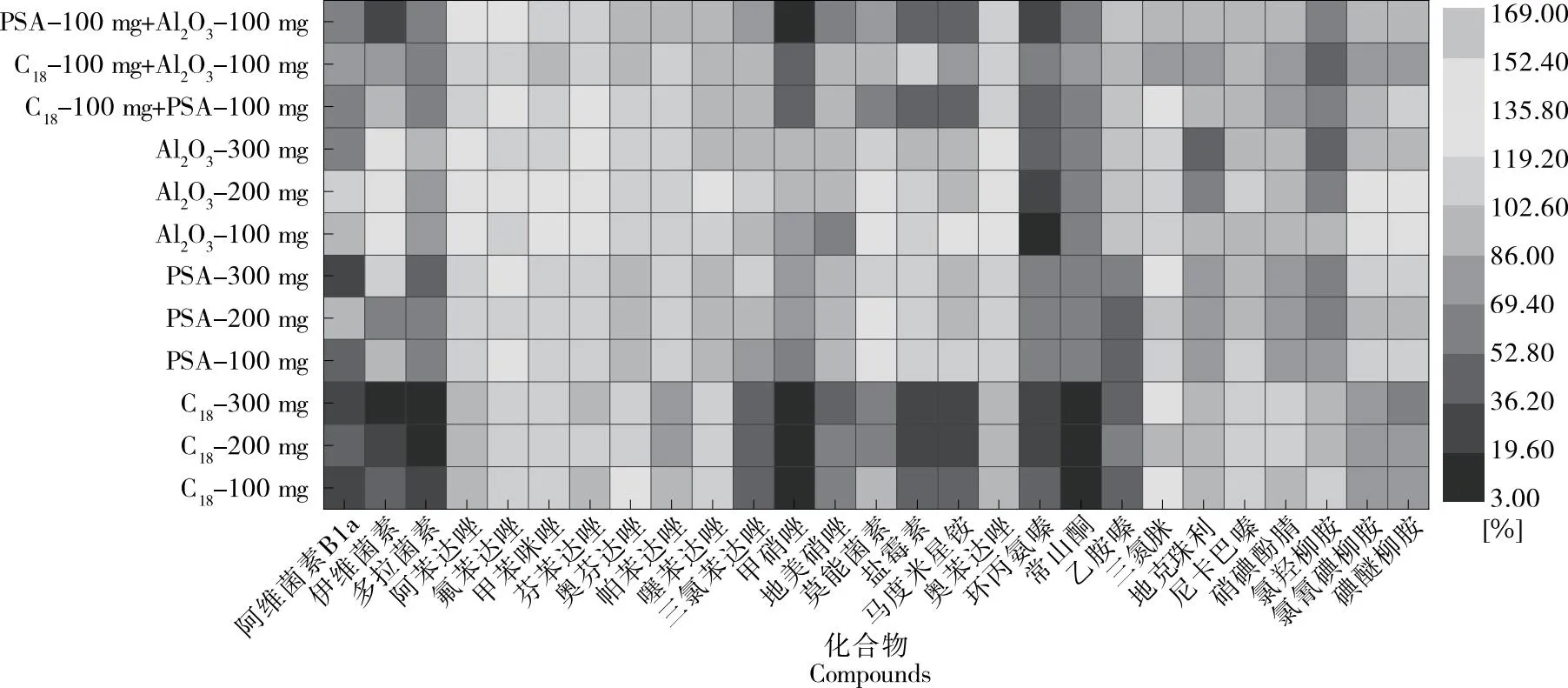
圖4 吸附劑對藥物提取回收率的影響
2.4 基質效應
在應用液相色譜串聯質譜特別是配有電噴霧離子源(ESI)對待測物進行分析時,進樣液中的雜質可能對分析物產生干擾,產生基質效應,嚴重影響方法的精密度、準確度,所以要對基質效應進行分析和評估[17]。基質效應消除或補償的辦法很多,常用基質匹配標準溶液校準方法[18]。研究涉及27種抗寄生蟲藥物在凡納濱對蝦基質中的定性定量分析測定,藥物種類較多,且基質中的蛋白、脂肪等雜質容易產生基質抑制或增強效應[19]。由圖5可知,馬度米星胺和環丙氨嗪顯示出基質增強效應,數值達到148.4%和128.8%,而其余25種化合物顯示出基質抑制效應,除硝碘酚腈外均小于80%。基質、前處理方法、化合物濃度都是影響基質效應的因素,為了抵消或補償基質效應帶來的影響,采取空白基質標液對線性關系、回收率、精密度等進行考察和實際樣品測定。
2.5 線性范圍、檢出限和定量限
用空白樣品提取液對27種抗寄生蟲藥物標準溶液進行稀釋,配制成0.5,1.0,2.0,5.0,10,50,100,200 ng/mL標準曲線系列并上機分析。以質量濃度為橫坐標,化合物定量離子峰面積為縱坐標進行線性回歸擬合,得到回歸方程和相應的皮爾森相關系數r[20]。方法檢出限(limit of detection,LOD)和定量限(limit of quantitation,LOQ)分別以3倍和10倍信噪比結合前處理過程中的稀釋倍數以及稱樣質量進行計算,結果見表2。
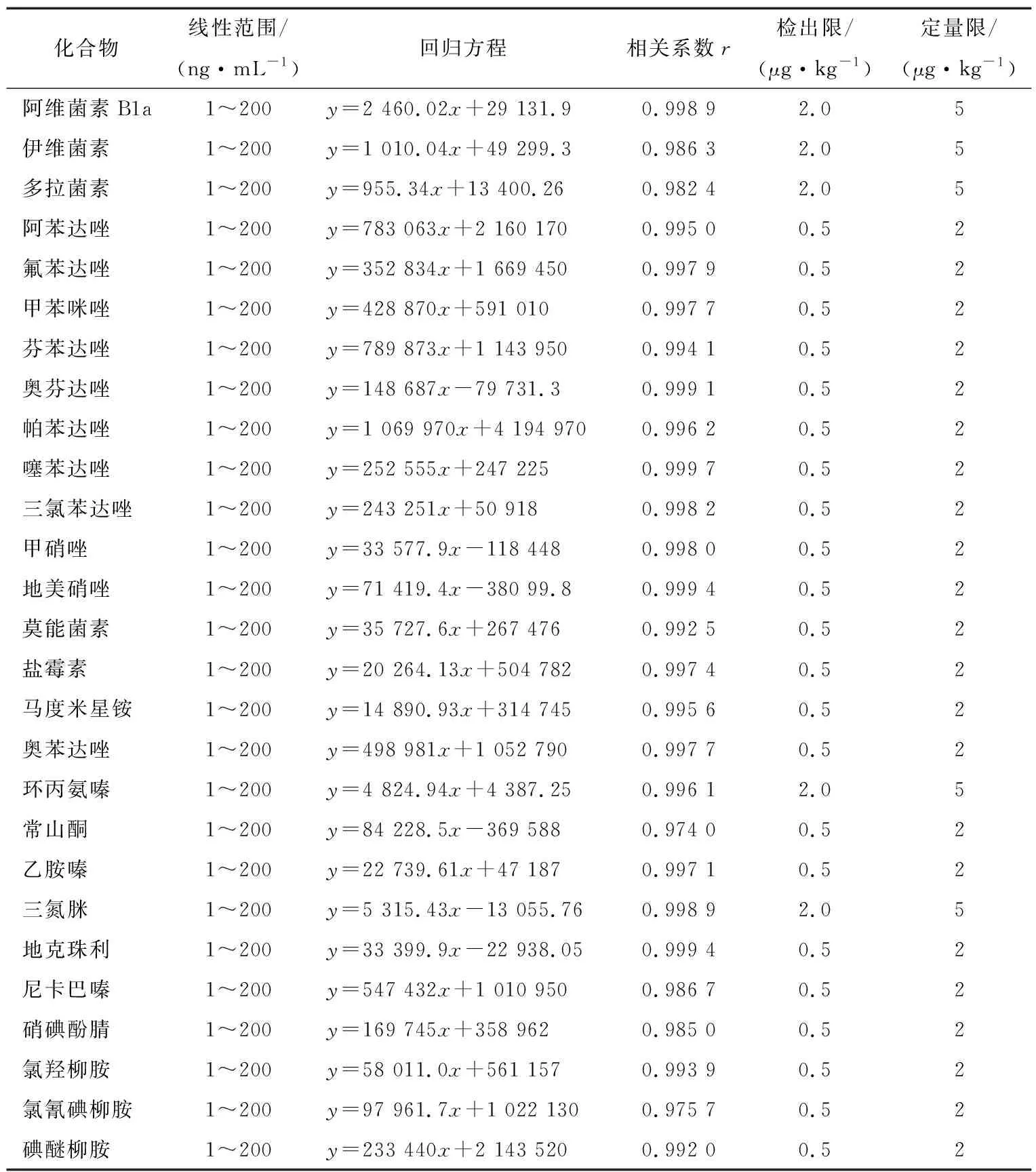
表2 抗寄生蟲藥物的線性范圍、回歸方程、相關系數以及檢出限和定量限
2.6 回收率和重復性
稱取凡納濱對蝦空白樣品2 g(精確至0.01 g)并進行標準溶液(1.3)空白基質添加(如表3所示),制備成低、中、高(5,10,50 μg/kg)3個水平的凡納濱對蝦陽性添加樣品,每個添加水平進行6次平行試驗。陽性添加樣品在處理前靜置0.5 h,然后按照1.4進行提取、凈化,上機分析后計算方法加標回收率和批內變異系數;重復3 d,以計算和分析批間變異系數,見表3。結果表明,方法回收率為61.90%~118.54%,批內變異系數為0.57%~12.56%,批間變異系數為1.29%~16.47%。
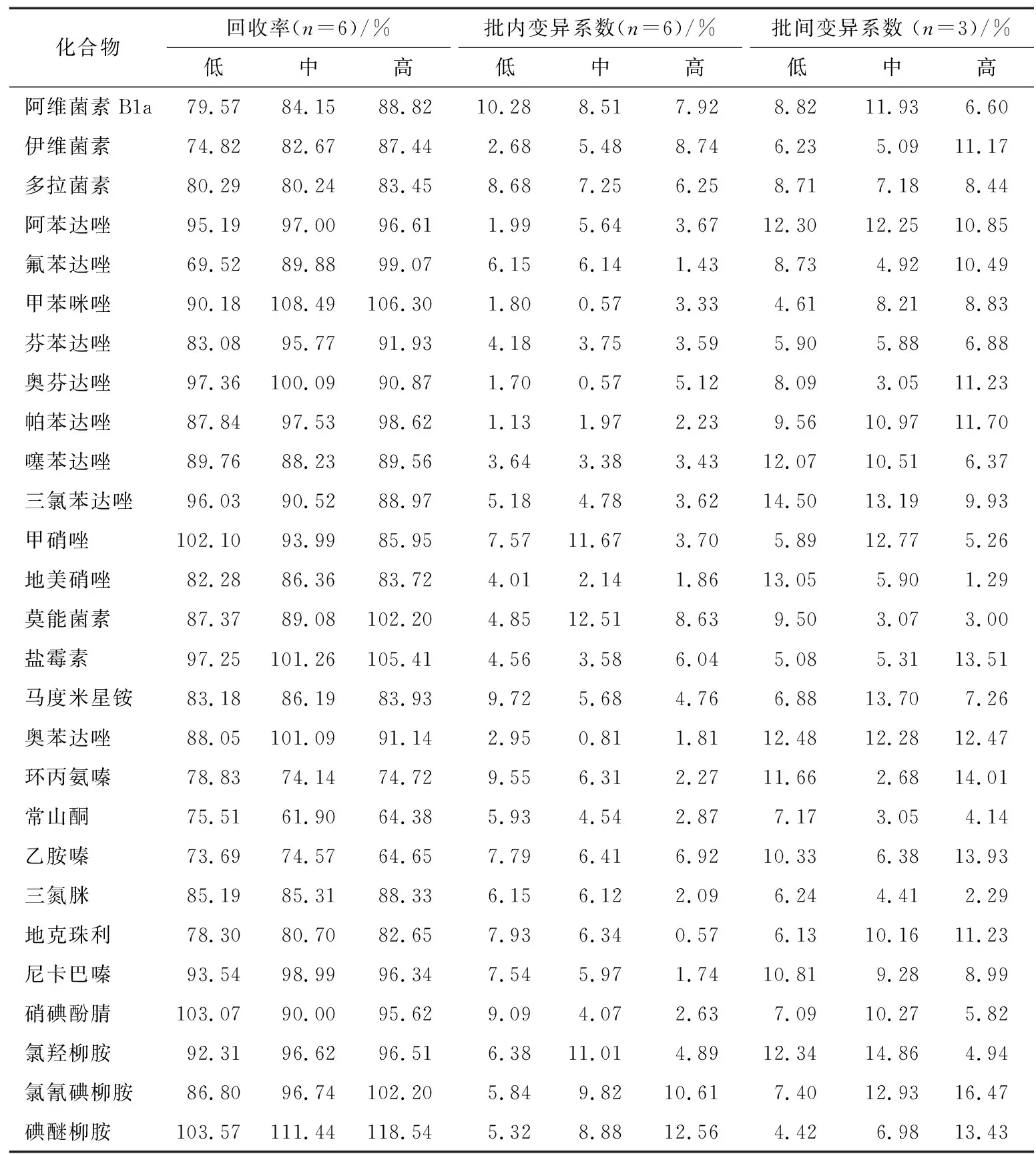
表3 凡納濱對蝦在不同添加水平下的回收率和變異系數
2.7 實際樣品測定
應用該方法對市場中購買的凡納濱對蝦樣品進行抗寄生蟲藥物殘留檢測。共測定凡納濱對蝦樣品56批,其中1批樣品有阿苯達唑檢出,1批樣品有地克珠利檢出,但均未達到該方法的定量限。阿苯達唑和地克珠利的檢出,說明已經有抗寄生蟲藥物滲透、污染到了南美白對蝦等水產品養殖環境中并在水產品中存在藥物存留的情況,數值雖然未達到該方法的定量限值,但也不應該忽視這一現實問題。這可能不是常規藥物施用而導致的藥物在飼養動物體內的藥物殘留問題,可能涉及到了畜產水產養殖、藥物遷移、養殖排棄物處置等系列問題,應予以關注。
3 結論
研究建立了液相色譜串聯質譜測定凡納濱對蝦中27種抗寄生蟲藥物的方法,藥物經提取、凈化后,于色譜柱進行分離,電噴霧離子源正負離子同步掃描,多反應監測模式測定。該方法前處理簡單、準確度高、精密度好。試驗研究的樣品基質為凡納濱對蝦,后續研究將繼續擴展蝦類品種和藥物殘留數量,以期適應多樣品基質種類和多藥物殘留的測定要求。
