基于重組蝦鐵蛋白裝載山奈酚的三種不同方法的比較分析
2023-12-29 08:21:08李蝶張斌申淼淼袁欣侯原菲李樹紅李冉張志清李美良
食品與發酵工業 2023年24期
李蝶,張斌,申淼淼,袁欣,侯原菲,李樹紅,李冉,張志清,李美良
(四川農業大學 食品學院,四川 雅安,625000)
山奈酚(kaempferol)是常見的典型天然多羥基黃酮類化合物,呈現出黃色粉末狀態,廣泛存在于各種水果、蔬菜及中草藥等天然植物中。具有抗癌[1]、抗氧化、抗炎、防治糖尿病[2]、保護神經、肝臟和心肌、抑制蛋白激酶等多方面的營養保健作用。但山奈酚水溶性差[3],導致其在食品營養及醫藥領域的生物利用率低,因此提高其水溶性和穩定性是解決應用受限的關鍵問題。目前,已有學者將山奈酚包裹成白蛋白納米粒[4]以及負載于殼聚糖內[3],有效提高了山奈酚的生物利用率。
鐵蛋白(ferritin)是一類廣泛存在于動物、植物及微生物中的儲鐵蛋白,主要由蛋白質外殼和鐵核兩部分組成[5],鐵蛋白外殼分子結構保守,內外徑分別為8 nm和12 nm,由24個亞基組成的中空籠狀結構[6]。因此鐵蛋白是一類天然的納米載體,剛性的蛋白質外殼使其在納米材料的應用研究中具有優勢。并且鐵蛋白分布廣泛、來源豐富、含量高、便于分離純化、無毒無害、生物相容性好,還是一類很好的營養物質。對于大腸桿菌誘導產生日本囊對蝦鐵蛋白(Marsupenaeusjaponicusferritin, MjF)具有無需脫鐵和可大批量生產的特點。目前,將食品生物活性小分子物質裝載進鐵蛋白腔內主要依賴于pH調控的可逆組裝特性,例如鐵蛋白分子裝載β-胡蘿卜素[7]、花色苷[8]、蘆丁[9]及姜黃素[10]等。然而,有學者研究表明,鐵蛋白在極酸性或極堿性條件下發生解離重組并非完全可逆[11],極酸極堿處理后的鐵蛋白的二級結構將遭到破壞。為避免此類情況發生,通過對鐵蛋白進行改性后再裝載的方法不斷涌現,超聲處理[12]、大氣冷離子體處理[13]、高靜壓處理[14]、脈沖電場處理[15]、高濃度尿素誘導[16]、熱處理[17]等改性方法,都使得鐵蛋白能在溫和的條件下進行解離。
超聲波技術已經廣泛應用于制備納米材料,例如乳液和含β-胡蘿卜素的酪蛋白納米顆粒[18]。并且張晨曦等[12]研究了超聲輔助法制備鐵蛋白-蝦青素復合物,采用超聲處理擴大鐵蛋白通道對蝦青素進行裝載。本文旨在對比超聲輔助法、高濃度尿素誘導法以及pH調控法3種不同裝載法制備的重組蝦鐵蛋白-山奈酚(recombinantMarsupenaeusjaponicusferritin kaempferol, rMjFK)復合物的各方面性質,探究出更溫和的裝載方法。
1 材料與方法
1.1 材料與試劑
含蝦重組鐵蛋白質粒(pET-3a)的大腸桿菌BL21(DE-3),中國農業大學;山奈酚(純度≥98%)、胰蛋白胨、酵母浸出粉、甘氨酸、考馬斯亮藍R-250、巰基還原劑、氫氧化鈉、尿素、硫酸銨、氯化鈉、十二烷基硫酸鈉、冰乙酸、甲醇、二甲基亞砜,北京索萊寶科技有限公司。
1.2 儀器與設備
蛋白純化系統、凝膠成像系統、電泳儀,美國Bio-Rad公司;透射電子顯微鏡,日本電子株式會社;納米粒度及Zeta電位儀,美國Malvern公司;靜音超聲波清洗器,昆山超聲儀器有限公司;傅里葉變換紅外光譜儀,美國Thermo公司。
1.3 實驗方法
1.3.1 重組蝦鐵蛋白(rMjF)的誘導表達與純化
本實驗對鐵蛋白純度要求較高,所以純化方法在馬貴紅等[19]的基礎上稍作修改。將含轉入日本囊對蝦重組鐵蛋白(rMjF)質粒(pET-3a)的大腸桿菌BL21(DE-3)接種于LB培養基[含50.0 μg/mL氨芐青霉素(ampicillin, AMP)]中,37 ℃,180 r/min條件下進行活化12 h。再擴培于含AMP(含50.0 μg/mL)的LB培養基中,37 ℃,180 r/min條件下進行培養至OD600達到0.6左右時,加入異丙基β-D-1-硫半乳糖苷(isopropyl β-D-1-thiogalactoside, IPTG)誘導表達8 h。
誘導后的菌液采用10 000 r/min離心5 min,棄去上清液,收集菌體并稱重。菌體重懸于25 mmol/L Tris-HCl(pH 7.5)緩沖液中,在經過凍融裂解后按料液比1∶500(g∶mL)加入溶菌酶(母液100 mg/mL)。超聲細胞破碎(功率300 W,超聲破碎20 min,開2 s,關4 s)20 min,將破碎后的菌液進行熱處理,于65 ℃水浴鍋內加熱15 min后離心(10 000 r/min,15 min)收集上清液。上清液提前放于4 ℃冰箱中降溫,再進行體積分數為20%~40%(先采用20%飽和硫酸銨處理,收集上清液后將硫酸銨的飽和度提升至40%,收集沉淀復溶)飽和硫酸銨復合處理。再將樣液置于300 kDa透析袋中于25 mmol/L Tris-HCl(pH 7.5)緩沖液中透析24 h,每8 h更換一次透析液,共3次,以便除去硫酸銨,超濾濃縮后4 ℃貯存備用。
將處理后的纖維素DE-52填料裝入弱陰離子交換柱(1.6 cm×20 cm)后,用25 mmol/L Tris-HCl(pH 7.5)緩沖液平衡柱子(1 mL/min)。將透析濃縮后的樣品上柱,按照設定的洗脫程序進行純化,蛋白純化系統的濾光片為280 nm。洗脫程序如下:
a)雜蛋白洗脫:Buffer A(25 mmol/L Tris-HCl,pH 7.5)100%,洗脫體積80 mL,流速1 mL/min;
b)梯度洗脫:Buffer A 90%,Buffer B(含NaCl的25 mmol/L Tris-HCl,pH 7.5)10%,洗脫體積80 mL,流速1 mL/min;Buffer A 85%,Buffer B 15%,洗脫體積80 mL,流速1 mL/min;Buffer A 70%,Buffer B 30%,洗脫體積80 mL,流速1 mL/min;
c)線性洗脫:Buffer A 70%~0%,Buffer B 30%~100%,洗脫體積80 mL,流速1 mL/min;
d)梯度洗脫:Buffer B 100%,洗脫體積80 mL,流速1 mL/min。將出峰部分收集,超濾濃縮后于-20 ℃貯存。用Bradford法(考馬斯亮藍試劑盒)測定蛋白濃度[20]。
1.3.2 rMjF的聚丙烯酰胺凝膠電泳分析
1.3.2.1 Native-PAGE電泳
將純化的蛋白溶液和天然樣品上樣緩沖液以體積比2∶1混勻,參照LAEMMLI[21]的方法進行Native-PAGE電泳,根據電泳后條帶判斷相應蛋白樣品的表觀分子質量。Native-PAGE濃縮膠和分離膠分別為6%GEL和5%GEL,凝膠板為80 mm×73 mm×1 mm,每個泳道點樣12 μL,Marker 10 μL。Native-PAGE電泳在4 ℃,恒流5 mA的條件下進行,待溴酚藍遷移線距離分離膠底部2 mm處電泳結束(約4 h)。電泳結束后,采用考馬斯亮藍R-250進行染色1 h并于脫色液中脫色。
1.3.2.2 SDS-PAGE電泳
SDS-PAGE電泳濃縮膠和分離膠分別為12%GEL和5%GEL。將純化的蛋白溶液和SDS-樣品上樣緩沖液以2∶1(體積比)的比例混勻,然后沸水浴煮7 min,接著每個泳道點樣12 μL,Marker 10 μL。SDS-PAGE電泳在室溫下進行,濃縮膠電壓80 V,分離膠電壓120 V,電泳完成后使用考馬斯亮藍R-250進行染色1 h,并于脫色液中脫色。
1.3.3 rMjF裝載山奈酚的制備
1.3.3.1 超聲輔助法制備rMjFK復合物
將山奈酚溶解于二甲基亞砜(dimethyl sulfoxide,DMSO)中,配制成1.0 μmol/L的山奈酚溶液。參考張晨曦等[12]的方法制備,將5 mL 0.1 μmol/L的rMjF樣品于35 ℃,100 W條件下預處理30 min后以摩爾比為1∶500加入1.0 μmol/L山奈酚溶液進行超聲60 min制備得到復合物。處理后的復合物樣液置于超濾離心管中除去游離的山奈酚,并在365 nm的吸收波長下測定游離山奈酚的吸光度,代入山奈酚標準曲線公式(y=0.072 4x+0.040 7,R2=0.999 2),計算出游離山奈酚的含量,從而確定山奈酚的裝載量,每個實驗重復3次。
1.3.3.2 尿素誘導法制備rMjFK復合物
參考YANG等[16]的方法制備,并稍作修改。用8 mol/L的尿素溶液誘導0.1 μmol/L的rMjF變性解離為單亞基,于恒溫磁力攪拌器下室溫攪拌25 min后,以1∶500的摩爾比緩慢加入1.0 μmol/L山奈酚溶液,繼續攪拌20 min。再用Tris-HCl緩沖液透析3次除去尿素以及游離山奈酚后制得復合物溶液,測定山奈酚的含量,從而確定山奈酚的裝載量,計算方法同1.3.3.1節,每個實驗重復3次。
1.3.3.3 pH調控法制備rMjFK復合物
將0.1 μmol/L的rMjF樣品調pH至2.0,于恒溫磁力攪拌器下攪拌30 min,后以1∶500的摩爾比緩慢加入1.0 μmol/L山奈酚溶液,繼續攪拌5 min,最后將pH調節至7.0。處理后的復合物樣液置于超濾離心管中除去游離的山奈酚,并測定游離山奈酚的含量,從而確定山奈酚的裝載量,計算方法同1.3.3.1節,每個實驗重復3次。
1.3.4 rMjFK復合物的表征
1.3.4.1 rMjFK復合物的聚丙烯酰胺凝膠電泳分析
參考1.3.2節的方法進行分析。
1.3.4.2 不同裝載方法對rMjFK復合物的裝載率影響
針對3種不同裝載方法所制備的復合物分別為超聲輔助法制備的rMjF-山奈酚復合物(rMjFK1)、尿素誘導法制備的rMjF-山奈酚復合物(rMjFK2)、pH過渡法制備的rMjF-山奈酚復合物(rMjFK3)。采用超濾杯離心出游離山奈酚,并在365 nm的吸收波長下測定游離山奈酚的吸光度,代入山奈酚標準曲線公式(y=0.072 4x+0.040 7,R2=0.999 2),經計算得出游離山奈酚的含量,推出裝載的山奈酚含量,從而計算出復合物中山奈酚的裝載率,計算如公式(1)所示:
裝載率/%=裝載后山奈酚的含量/總rMjF的含量
(1)
1.3.4.3 rMjFK復合物的透射電子顯微鏡(transmission electron microscope, TEM)分析
TEM制樣過程參考JI等[22]的方法。首先,取10 μL的樣品溶液(1 mg/mL)置于干凈的封口膜上,將碳膜包被的銅網倒扣在液滴上10 min,再用濾紙吸去多余的液體,然后用2%磷鎢酸進行靜置負染色10 min,最后用濾紙吸去多余染色液。等干燥后使用JEM-2000透射電鏡以100 kV在50 nm比例尺下觀察并拍攝結構成像。
1.3.4.4 rMjFK復合物的粒徑與電勢分析
采用Zetasizer Nano ZS納米粒度儀對樣品溶液進行測定,測試參考LI等[23]的方法并稍作修改。具體操作如下:將樣品液過0.45 μm的水系濾膜(減少大顆粒和氣泡的干擾),測定溫度為25 ℃,散射光角度為90°,測量時間為120 s,進行重復3次測量后取平均值進行分析,粒徑和電勢數據導出后采用Origin作圖。
1.3.4.5 rMjFK復合物的傅里葉紅外光譜分析
利用傅立葉紅外光譜儀測定rMjF以及復合物的二級結構,樣品溶液經透析后進行冷凍干燥,取1 mg與充分干燥的KBr顆粒放入研缽中,充分混合。將研磨好的混合粉末放入模具內制成的樣品薄片。將充分干燥后的KBr用同樣方法處理后作空白對照。紅外光譜儀分辨率4 cm-1,掃描次數為32次。
2 結果與分析
2.1 rMjF的純化與表征
2.1.1 rMjF的誘導表達
將含rMjF的大腸桿菌培養至細菌細胞濃度達到OD600為0.6時,添加IPTG進行誘導表達,通過變性電泳SDS-PAGE對含有rMjF誘導前后的菌體進行檢測。結果如圖1所示,泳道1為誘導表達前的菌液,泳道2為誘導表達后的菌液。觀察發現泳道2在20 kDa處出現一條特異性蛋白條帶,誘導后該蛋白條帶濃度明顯增加,且分子質量大小符合鐵蛋白亞基分子質量,證明目的蛋白誘導成功。
2.1.2 rMjF的分離純化
對誘導后的菌體進行分離純化,根據鐵蛋白具有良好的熱穩定性及蛋白鹽析的特性,對誘導表達后的菌體進行65 ℃熱變性和20%~40%硫酸銨分級鹽析進行初步分離。再采用DE-52弱陰離子交換柱層析進一步純化。由圖2-a可知,層析過程有4個蛋白峰,分別為峰Ⅰ、Ⅱ、Ⅲ、Ⅳ。經鑒定峰Ⅰ為雜蛋白峰,峰Ⅱ中含有少量雜蛋白,而峰Ⅲ幾乎不含雜蛋白,峰Ⅳ中含有大量雜蛋白。故取峰Ⅲ的蛋白做后續實驗。圖2-b是rMjF初步分離及層析純化過程的SDS-PAGE電泳圖,泳道1為菌液,可以看出其含有大量雜蛋白;泳道2對應的是65 ℃熱變性后的上清液,可以看出大量熱敏性雜蛋白被除去;泳道3和泳道4是經20%~40%分級鹽析處理后的條帶,可看出樣品還含有少量的雜蛋白;泳道5為經交換柱層析后的峰Ⅲ,此時樣品已經達到分析純,條帶中幾乎不含雜蛋白。
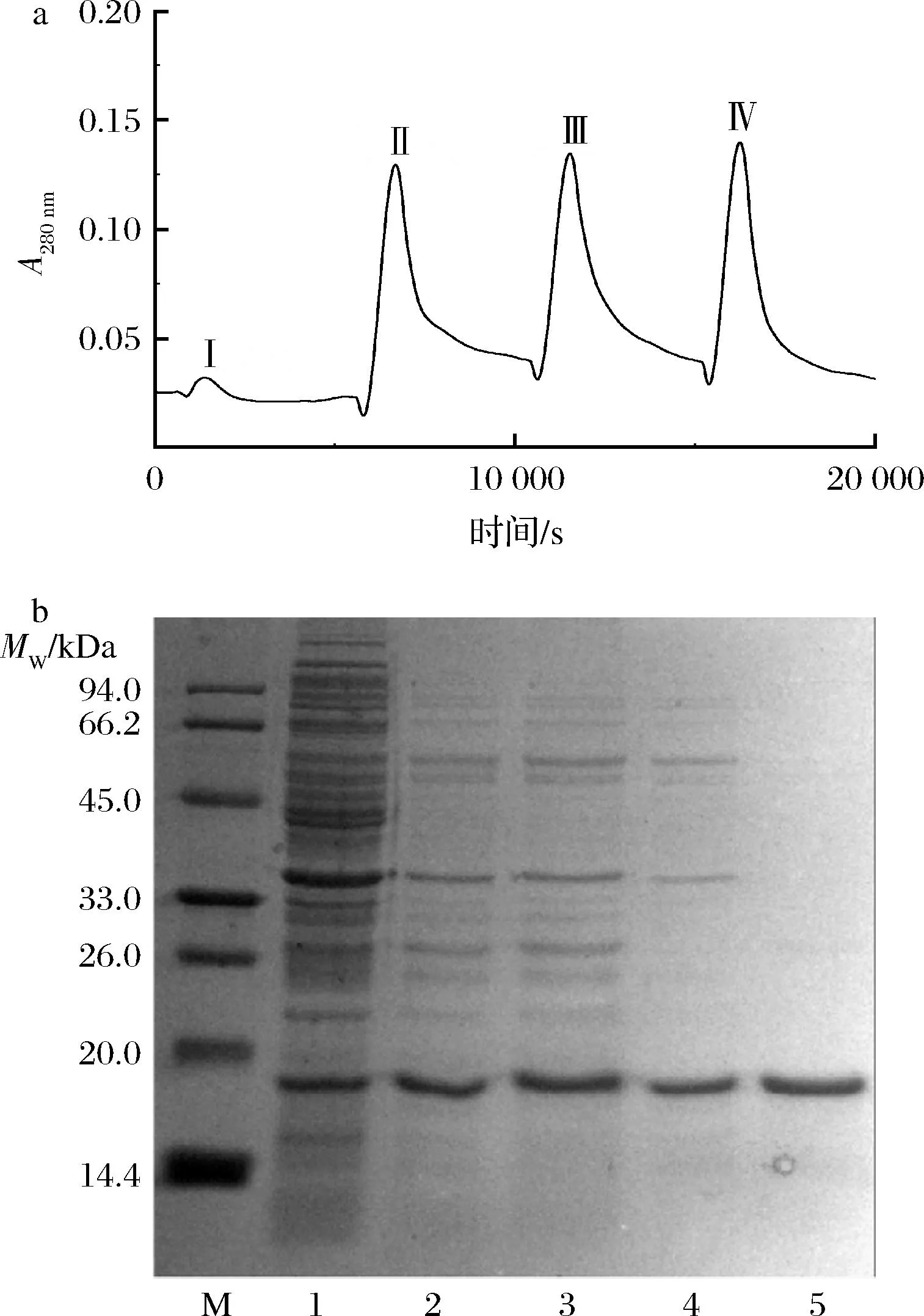
a-rMjF純化層析圖譜;b-rMjF純化過程SDS-PAGE電泳圖 泳道M-標準蛋白Marker(14.4~94.0 kDa);泳道1-菌液; 泳道2-經65 ℃水浴處理后的上清液;泳道3~4-硫酸銨分級 處理后的樣品;泳道5-層析純化后的峰Ⅲ圖2 rMjF分離純化過程中的層析圖譜與SDS-PAGE電泳圖Fig.2 Chromatographic and SDS-PAGE electrophoresis of rMjF during its purification process
2.1.3 rMjF的純度與亞基鑒定
對層析后的蛋白進行Native-PAGE電泳分析,結果如圖3-a,泳道1為rMjF,可明顯看出在440~232 kDa出現了一條較明顯的條帶,分子質量大小與WANG等[24]的研究結果一致。但在該條帶上方還有一條淺淺的條帶,這是由于蛋白濃度較高時會產生二聚體條帶于669 kDa處,與夏小雨等[25]的研究結果一致,由此可說明該蛋白已經達到電泳純。圖3-b為rMjF的SDS-PAGE電泳圖,泳道1同樣為純化后的rMjF,該泳道可明顯看出只有一個條帶,說明rMjF由單一亞基M亞基構成。經Quantity One軟件分析結果如圖3-c、圖3-D所示,得到rMjF的M亞基分子質量為18.4 kDa,該結果與ELVITIGALA等[26]所描述的M亞基分子質量一致。
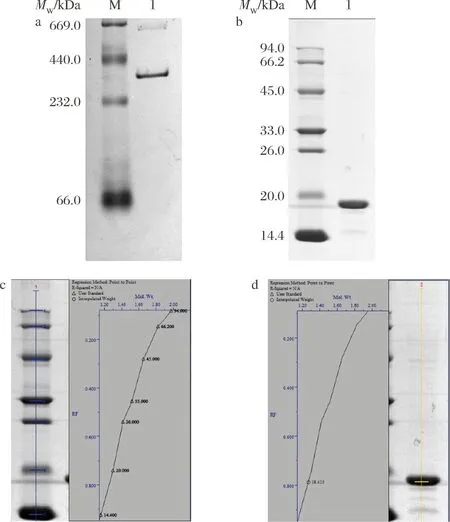
a-rMjF的Native-PAGE電泳圖;b-rMjF的SDS-PAGE電泳圖; c-Marker分子質量參考圖;d- rMjF分子質量定量圖圖3 rMjF的Native-PAGE和SDS-PAGE電泳圖Fig.3 Native-PAGE and SDS PAGE electrophoretic diagrams of rMjF注:a中的泳道M-標準蛋白Marker(669.0~66.0 kDa),泳道1-rMjF; b中的泳道M-標準蛋白Marker(14.4~94.0 kDa),泳道1-rMjF。
2.2 三種不同裝載方法制備的rMjFK復合物的表征
2.2.1 rMjFK復合物的聚丙烯酰胺凝膠電泳分析
采用3種不同裝載方式制備的復合物經透析除掉游離山奈酚后進行Native-PAGE以及SDS-PAGE電泳檢測,結果如圖4所示。泳道1~4分別為rMjF以及rMjF1、rMjF2、rMjF3,其亞基分子質量與蛋白分子質量與裝載前的rMjF一致,表明超聲處理、高濃度尿素誘導以及強酸性環境對亞基以及蛋白總分子質量影響不大。
2.2.2 不同裝載方法對復合物裝載率的影響
利用rMjF經超聲以及高濃度尿素處理后通道尺寸變大的特性和在pH 2.0~7.0轉變下的可逆自組裝特性,制備了3種復合物,分別為rMjFK1、rMjFK2和rMjFK3。復合物經超濾杯除掉游離山奈酚后,檢測游離山奈酚的含量,從而得出復合物的裝載率。結果如圖5所示,超聲輔助法所制備的復合物的裝載率最高,達到了20%;而尿素誘導法制備的復合物裝載率為18.5%;pH調控制備的復合物裝載率為16.7%,顯著低于超聲輔助法所制備的復合物裝載率。這是因為超聲結束后鐵蛋白通道會自動縮小,而尿素誘導鐵蛋白后需進行透析除去尿素,鐵蛋白通道才會慢慢縮小,這一過程是降低裝載率的主要因素。而pH調控鐵蛋白自組裝,這一過程具有隨機性,裝載率大小與摩爾比有關。
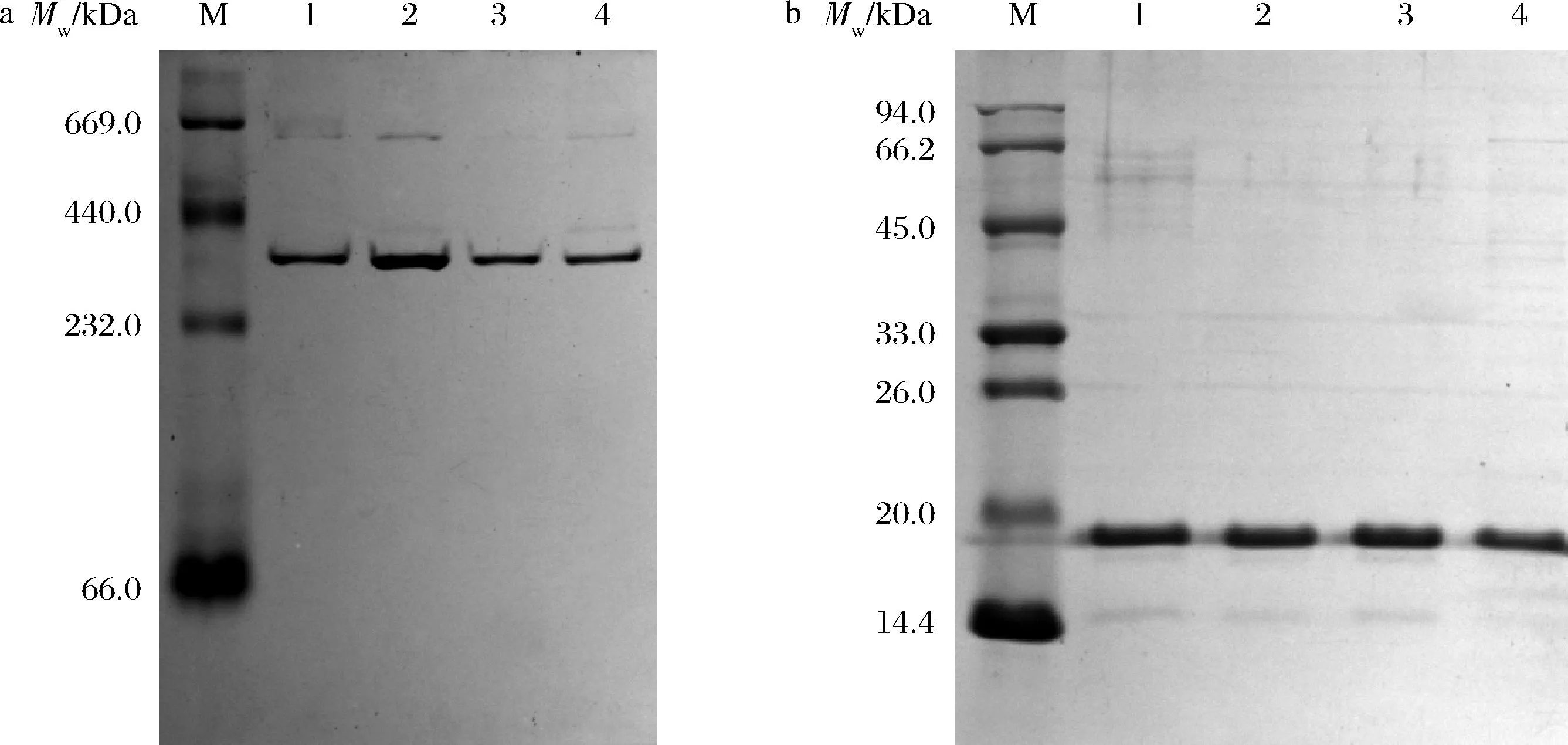
泳道M-標準蛋白Marker;泳道1-rMjF;泳道2-超聲輔助法制備的rMjFK1;泳道3-尿素誘導法制備的rMjFK2;泳道4-pH調控法制備的rMjFK3 a-rMjF與3種復合物的Native-PAGE電泳圖;b-rMjF與3種復合物的SDS-PAGE電泳圖圖4 rMjF與3種復合物的Native-PAGE和SDS-PAGE電泳圖Fig.4 Native-PAGE and SDS PAGE electrophoretic diagrams of rMjF and three compounds
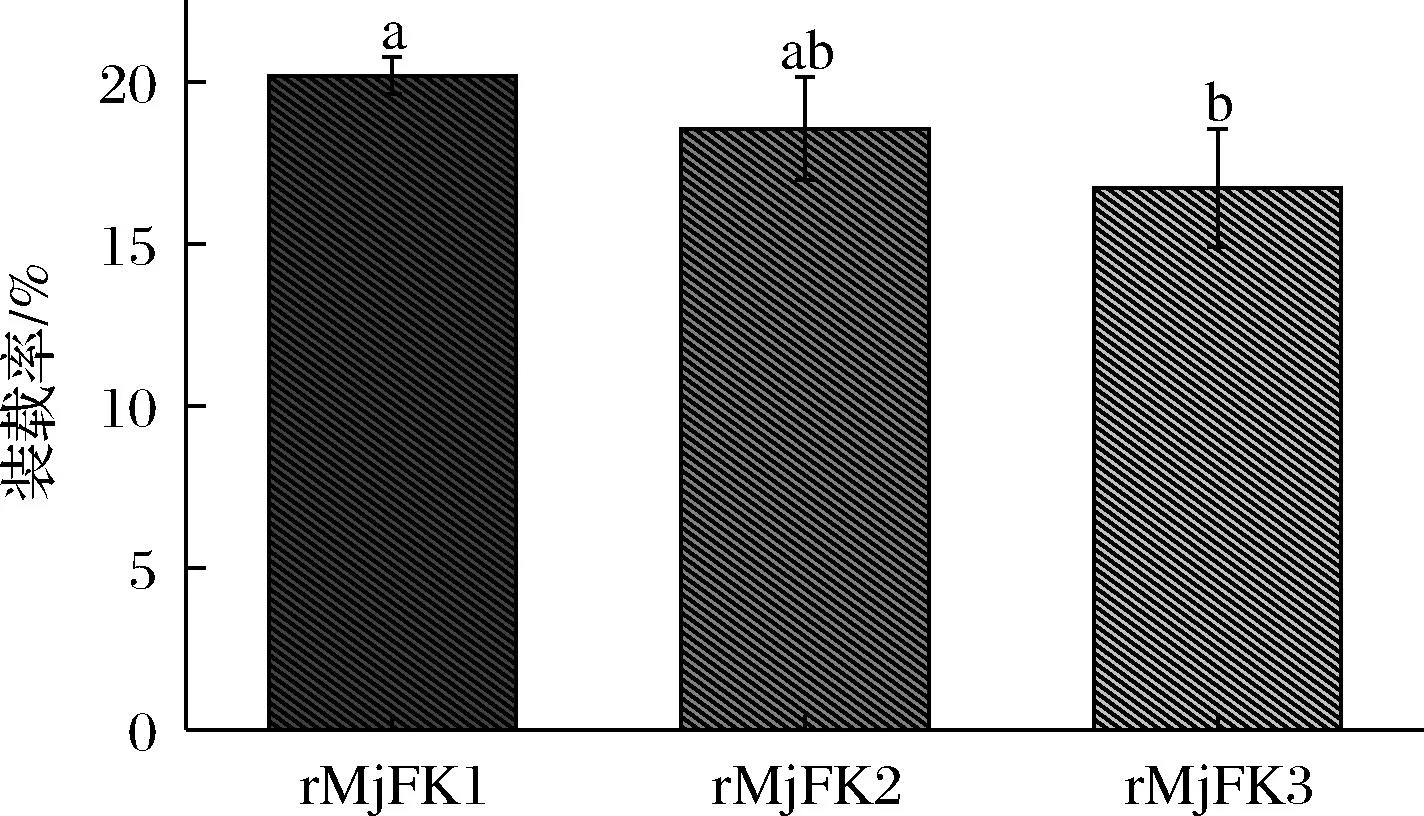
圖5 三種復合物裝載率的比較Fig.5 Comparison of loading rates of three compounds注:不同字母表示差異顯著(P<0.05)(下同)。
2.2.3 rMjFK復合物的TEM分析
圖6為rMjF與3種復合物的TEM圖,圖6-a為裝載前的rMjF,其中蛋白腔內有黑色的鈾核,這是由于在對鐵蛋白進行負染時鈾離子進入到了蛋白質內腔形成的[27]。而圖6-b~圖6-d分別為3種復合物的TEM圖,其中大部分蛋白腔內的黑色鈾核消失了。這是因為當化合物被裝載進鐵蛋白內部空腔,會限制乙酸鈾酰的進入與成核。所以山奈酚進入鐵蛋白內腔中占據內部空間,從而阻止了鈾離子進入腔內。圖6-b~圖6-d 3張電鏡圖中的部分鐵蛋白內部空腔呈現白色,證明山奈酚成功裝載進了rMjF的內部空腔[28]。其次由電鏡圖也可看出鐵蛋白在高濃度尿素以及強酸性環境中的聚集情況更為嚴重,而在超聲處理后的鐵蛋白只有較少的聚集,這是因為超聲波具有空化作用,能使較大的聚集體分散[29-30]。同時透射電鏡結果也可說明裝載后的鐵蛋白均保持了與裝載前的鐵蛋白一致的球型結構。
2.2.4 rMjFK復合物的粒徑與電勢分析
粒徑結果同樣可證明,采用馬爾文粒度儀測定了3種復合物的粒徑。粒徑分布結果如圖7所示,裝載前的鐵蛋白粒徑分布于13.54 nm左右,并且具有單一峰,說明溶液中不含有雜蛋白且粒徑較為分散。這與前人所描述的典型鐵蛋白尺寸大小相符[6]。pH調控法制備的復合物粒徑與其一致,而超聲輔助法和尿素誘導法制備的復合物粒徑大于裝載前的鐵蛋白粒徑。這是因為超聲和尿素處理都會導致鐵蛋白通道擴大,一旦停止,鐵蛋白通道會慢慢縮小,并不會完全恢復。同時與電鏡結果一致的是裝載后的蝦重組鐵蛋白具有不同程度的聚集情況,由圖7可知,裝載前的鐵蛋白具有單一峰,而3種復合物具有2~3個峰。如表1所示,超聲處理后的聚集體僅占29.1%,而尿素誘導和pH調控所產生的聚集體分別高達55.7%和66.2%。超聲輔助制備的物具有使粒徑統一的特點。
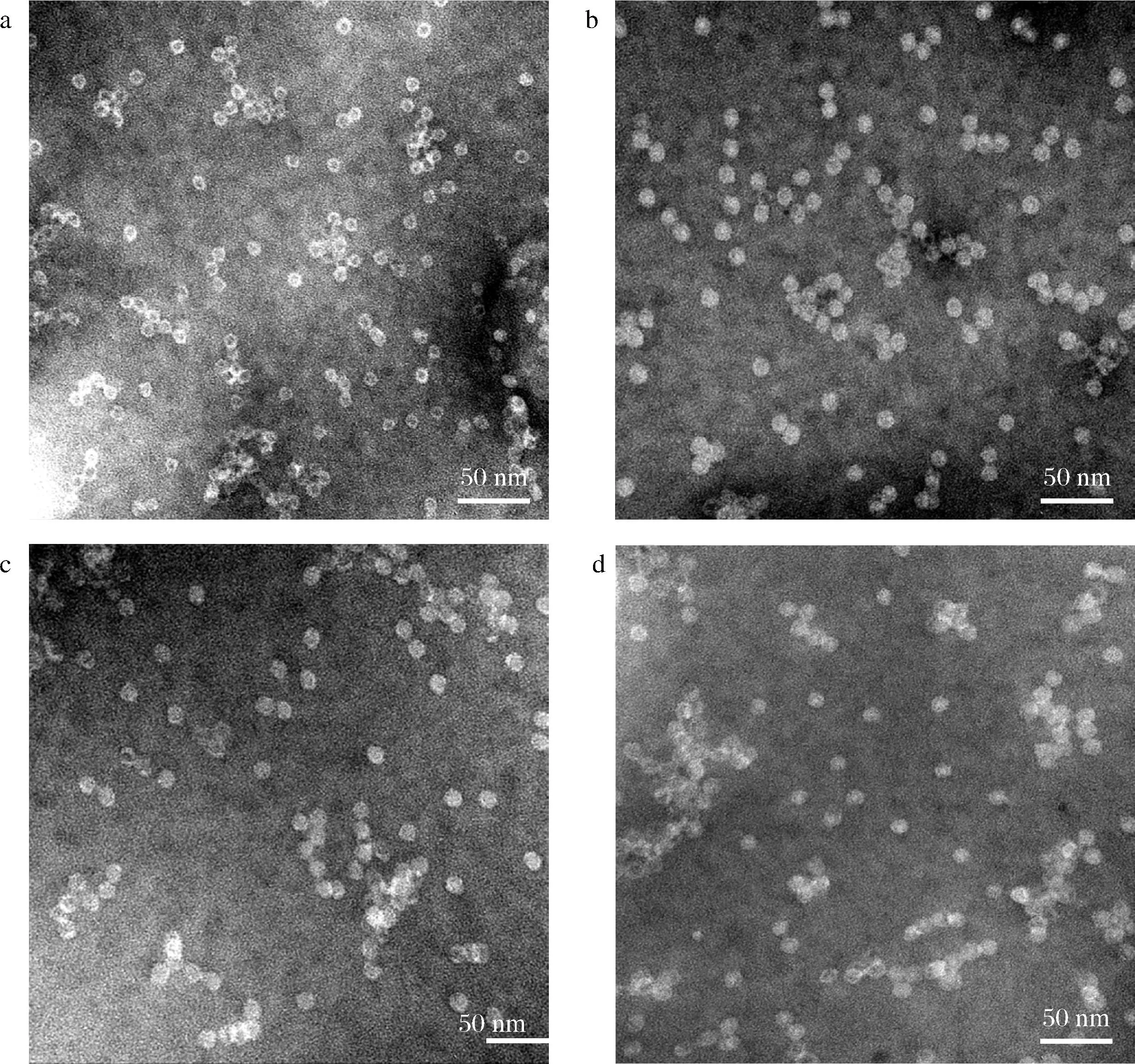
a-rMjF的TEM圖;b-rMjF1的TEM圖;c-rMjF2的TEM圖;d-rMjF3的TEM圖圖6 rMjF與3種復合物的TEM圖Fig.6 TEM images of rMjF and three compounds
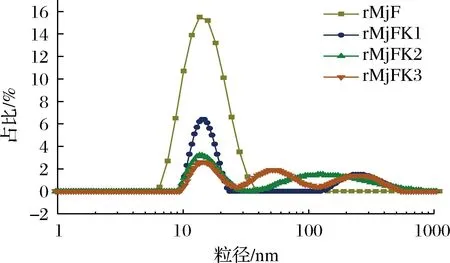
圖7 rMjF與3種復合物的粒徑分布圖Fig.7 Particle size distribution of rMjF and three compounds
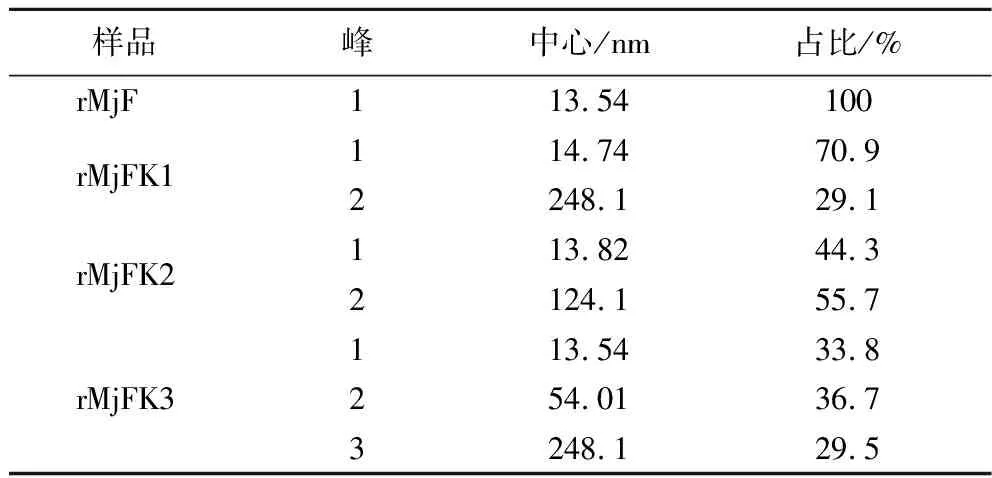
表1 rMjF與3種復合物的粒徑分布表Table 1 Particle size distribution table of rMjF and three compounds
Zeta電位一般用于評價或預測微粒分散體系的物理穩定性,一般Zeta電位絕對值越高,其粒子間的靜電斥力也越大,物理穩定性也就越好[31]。結果如圖8所示,其中超聲輔助法制備的復合物電位與處理前鐵蛋白電位結果沒有顯著差異,而尿素誘導法和pH調控法制備的復合物的電位結果與處理前鐵蛋白電位結果具有顯著差異。該結果與粒徑結果一致,超聲處理對蛋白質聚集有抑制作用。
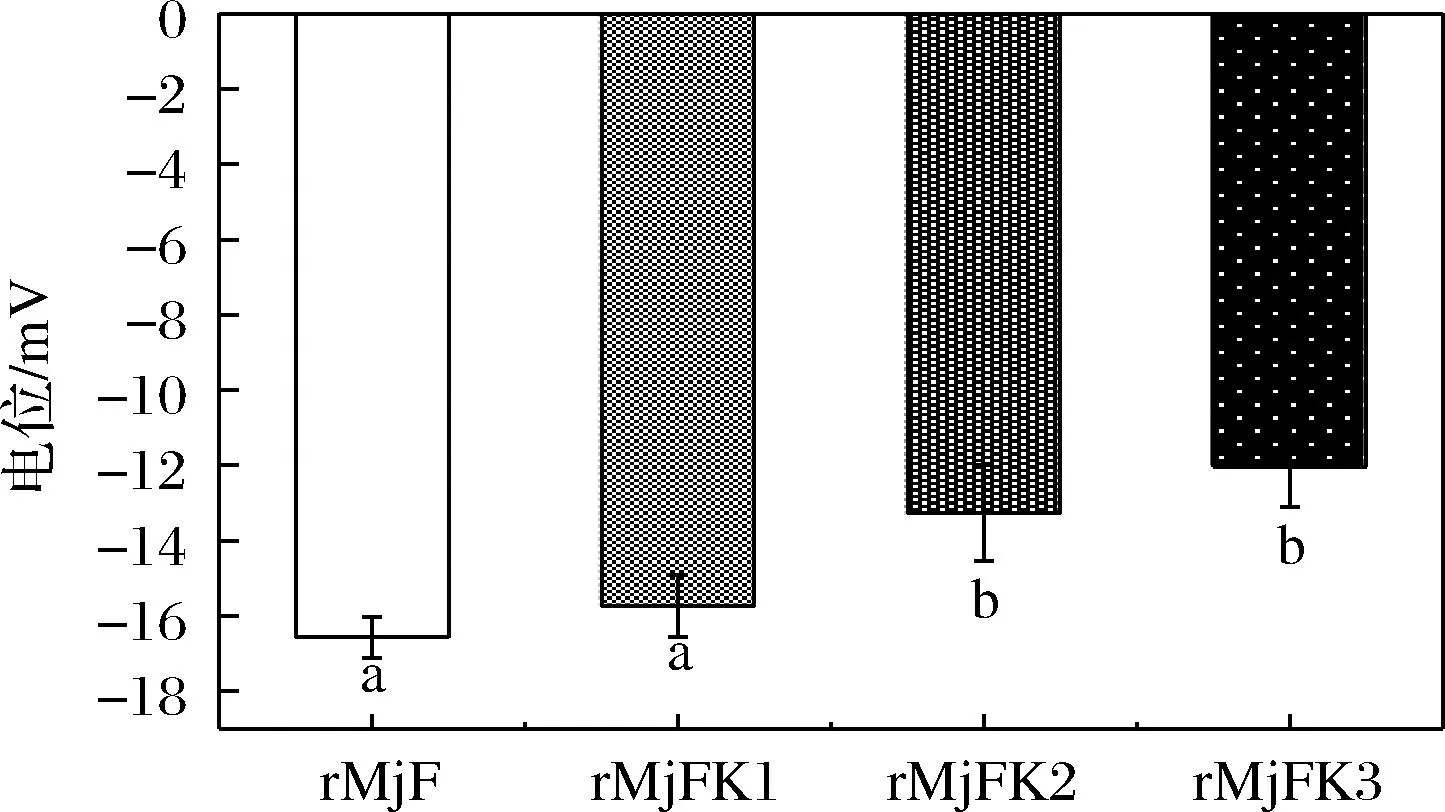
圖8 rMjF與3種復合物的Zeta電位柱形圖Fig.8 Zeta potential histogram of rMjF and three compounds
2.2.5 rMjFK復合物的傅里葉紅外光譜分析

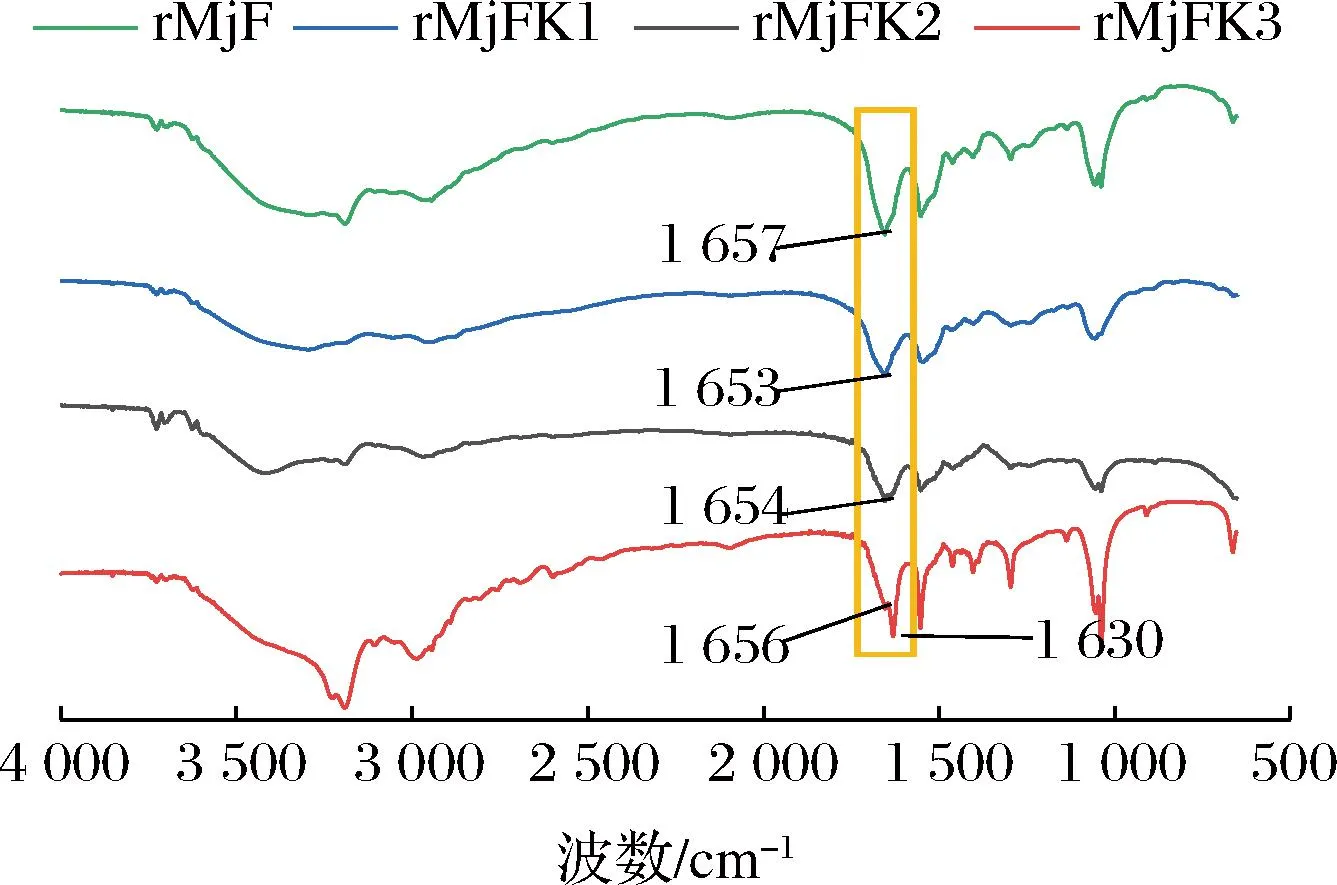
圖9 rMjF與3種復合物的傅里葉紅外光譜圖Fig.9 Fourier infrared spectra of rMjF and three compounds
3 結論與討論
本文采用超聲輔助、尿素誘導以及pH調控3種不同的裝載方法制備了rMjFK復合物。與傳統的通過pH調控使鐵蛋白在極酸極堿環境中自組裝裝載活性小分子物質所對比,超聲輔助法裝載活性小分子物質具有以下優點:相同摩爾比條件下,所制備的復合物裝載率更高;能有效抑制鐵蛋白的聚集情況,制備的復合物具有較好的分散性;可使鐵蛋白在溫和的環境中進行裝載,避免二級結構遭到破壞。而尿素誘導雖然不對鐵蛋白二級結構產生嚴重破壞,但其裝載過程耗時長,成本高,且裝載率低于超聲輔助法。所以超聲輔助法制備鐵蛋白與水溶性差的活性小分子的復合物具有良好的應用前景。
