骨髓間充質干細胞衰老研究進展
2024-01-12 13:13:46于淼瑛宋曉東王艷輝劉曾旭
中國骨質疏松雜志 2023年12期
于淼瑛 宋曉東 王艷輝 劉曾旭
1.上饒師范學院生命科學學院,江西 上饒 334001 2.江西醫學高等專科學校基礎醫學院,江西 上饒 334000 3.南昌大學基礎醫學院,江西 南昌 330006
多種損傷或應激刺激均可導致細胞衰老,產生表觀遺傳、形態和功能改變,以及由多種促炎因子組成的衰老相關分泌表型(senescence-associated secretory phenotype,SASP),是各種年齡相關和代謝性疾病的驅動因素[1-2]。間充質干細胞(mesenchymal stem cells,MSCs)具有組織再生、抗炎和免疫調節等功能,且不受免疫排斥、惡性轉化和倫理問題等限制;同時,易于分離和擴增,被認為是最適合移植治療的干細胞類型。目前,骨髓組織是MSCs最重要的獲取來源,骨髓間充質干細胞(bone marrow mesenchymal stem cells,BM-MSCs)已在多種創傷性、炎癥性、自身免疫性和退行性疾病的研究與治療中被廣泛應用且效果良好[3]。隨年齡增長而出現的自然衰老和由疾病損傷等原因造成的復制性與應激性衰老導致BM-MSCs的形態學、染色質結構、遷移歸巢、再生分化、代謝平衡和旁分泌等細胞特性發生顯著改變,造成其有益功能急劇下降并誘發慢性炎癥。衰老BM-MSCs的積累造成骨髓的更新活性、新骨形成率和骨折修復率顯著降低,引發骨骼愈合延遲、骨丟失和骨關節炎等骨組織退行性病變,是骨質疏松癥等代謝性骨病和高齡體重增加以及機體衰老的核心誘因。為滿足細胞移植的數量需求而對分離后BM-MSCs進行的長期體外擴增可誘發其復制性與應激性衰老,導致移植后的組織修復和免疫調控能力下降,且極易引起免疫排斥、炎癥反應和惡性轉化等安全風險[4-5](圖1)。因此,全面探討和深入理解BM-MSCs的衰老過程是延緩和逆轉年齡相關退行性病變以及完善BM-MSCs臨床治療的重要前提。
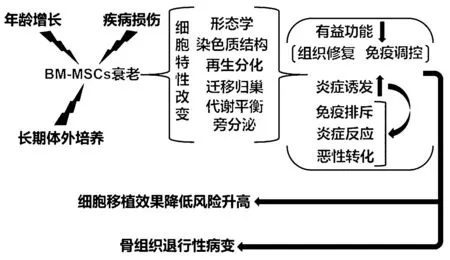
圖1 BM-MSCs衰老導致骨組織退行性病變和移植價值降低Fig.1 Senescence of BM-MSCs leads to bone tissue degenerative diseases and decreased transplantation value
1 衰老改變BM-MSCs細胞特性
1.1 衰老導致BM-MSCs出現細胞周期阻滯
衰老BM-MSCs中,多能性轉錄因子和細胞周期調控基因的表達發生顯著變化,引起細胞周期G1/G0期阻滯和S期縮短[4,6]。Cdkn2a(Cyclin-dependent kinase 2a)位點編碼的腫瘤抑制因子p16INK4A、成視網膜細胞瘤蛋白(retinoblastoma,Rb)、腫瘤抑制基因p53及其下游因子p21WAF1/CIP1、以及共濟失調毛細血管擴張癥突變蛋白(ataxia-telangiectasia mutated,ATM)是經典的細胞周期抑制因子,其活性表達在BM-MSCs衰老過程中顯著增加。各種刺激下的應激和持續過度的DNA損傷反應(DNA damage response,DDR)通過激活p16INK4A-Rb或p53-p21WAF1/CIP1信號通路誘導細胞進入永久性的細胞周期阻滯和顯著的結構功能改變是衰老細胞的共同特征;而消融p16INK4A或p21WAF1/CIP1可逆轉BM-MSCs衰老[7-9]。快速衰老的MSCs中,p16INK4A、p53、p21WAF1/CIP1和ATM的表達均顯著增加;而延遲衰老的MSCs中p16INK4A表達上調,p21WAF1/CIP1、p53和ATM表達降低[10-11]。
MSCs的快速增殖是通過維持Nanog、Oct4、Sox-2、PBX1和Rex1等多能性轉錄因子表達以抑制p16INK4A和p21WAF1/CIP1等細胞周期抑制因子實現的。Nanog和Oct4直接結合到DNA甲基轉移酶DNMT1的啟動子上增強其表達以維持p16INK4A和p21WAF1/CIP1啟動子的DNA甲基化進而抑制兩者表達[12]。PBX1激活Nanog啟動子并與Nanog、Oct4和Sox-2合作抑制MSCs衰老,其下游通路包括通過抑制p16INK4A和p21WAF1/CIP1,以及AIF、cleaved caspase 3和CytC等凋亡蛋白的表達,并激活PI3K-AKT信號通路,以維持MSCs的自我更新和分化平衡;同時,通過減少活性氧(reactive oxygen species,ROS)積累,并調節PARP1、Ku70、Ku80和Rad51等DNA損傷修復蛋白表達,來降低ROS介導的DDR[13-14](圖2)。PI3K-AKT信號通路對MSCs衰老的調節具有雙重作用,AKT激活可降低p16INK4A、p53和p21WAF1/CIP1表達;而持續的AKT過度活化則直接磷酸化p21WAF1/CIP1觸發MSCs的過早衰老[15]。快速衰老的BM-MSCs中,Nanog、Oct4、Sox-2和Rex1的表達水平均顯著下降;而延遲衰老過程中,Sox-2和Rex1的表達水平沒有顯著變化[12-14]。
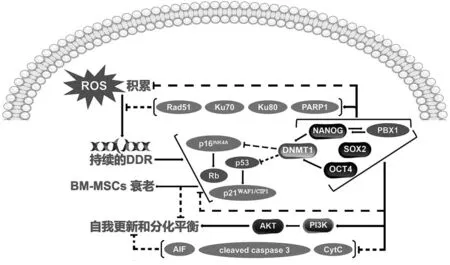
圖2 多能性轉錄因子抑制BM-MSCs衰老并促進其自我更新和分化平衡Fig.2 Pluripotent transcription factors inhibit senescence of BM-MSCs and promote their self-renewal and differentiation balance
1.2 衰老過程中BM-MSCs的表觀遺傳修飾改變
1.2.1衰老過程中BM-MSCs的染色質結構改變:衰老BM-MSCs中,染色質上出現大量密集緊縮轉錄不活躍的衰老相關異染色質灶(senescence-associated heterochromatin foci,SAHF),結構富含γH2AX和53BP1等DDR相關蛋白,且有利于維持BM-MSCs衰老所需的持續DNA損傷修復過程,抑制E2Fs活性及其調控的Cyclin A、Cyclin E、PCNA和MCM4等細胞增殖基因表達。同時,組蛋白去乙酰化酶(histone deacetylases,HDACs)表達降低,導致組蛋白的H3K9和H3K14乙酰化加劇,引起Naong、Oct4、Sox-2和端粒酶催化亞基Tert表達降低,以及眾多microRNAs表達激活來抑制高遷移率族蛋白A2(high mobility group A2,HMGA2)翻譯。HMGA2可激活Cyclin A、Cyclin E1、Cyclin F和CDC25A等細胞增殖基因,誘導PI3K-AKT與mTOR-p70S6K信號通路,并抑制p16INK4A等衰老基因表達[16-17]。DNA甲基轉移酶(DNA methyltransferases,DNMTs)通過促進組蛋白甲基轉移酶EZH1和EZH2增加p16INK4A和p21WAF1/CIP1啟動子區域的抑制性靶向標記。MSCs衰老過程中,DNMT1和DNMT3B表達逐漸下調,引起p16INK4A和p21WAF1/CIP1的表達升高(圖3)。與DNMT1和DNMT3B相反,DNMT3A在MSCs衰老過程中表達增加并誘導相關基因的新甲基化[18-19]。
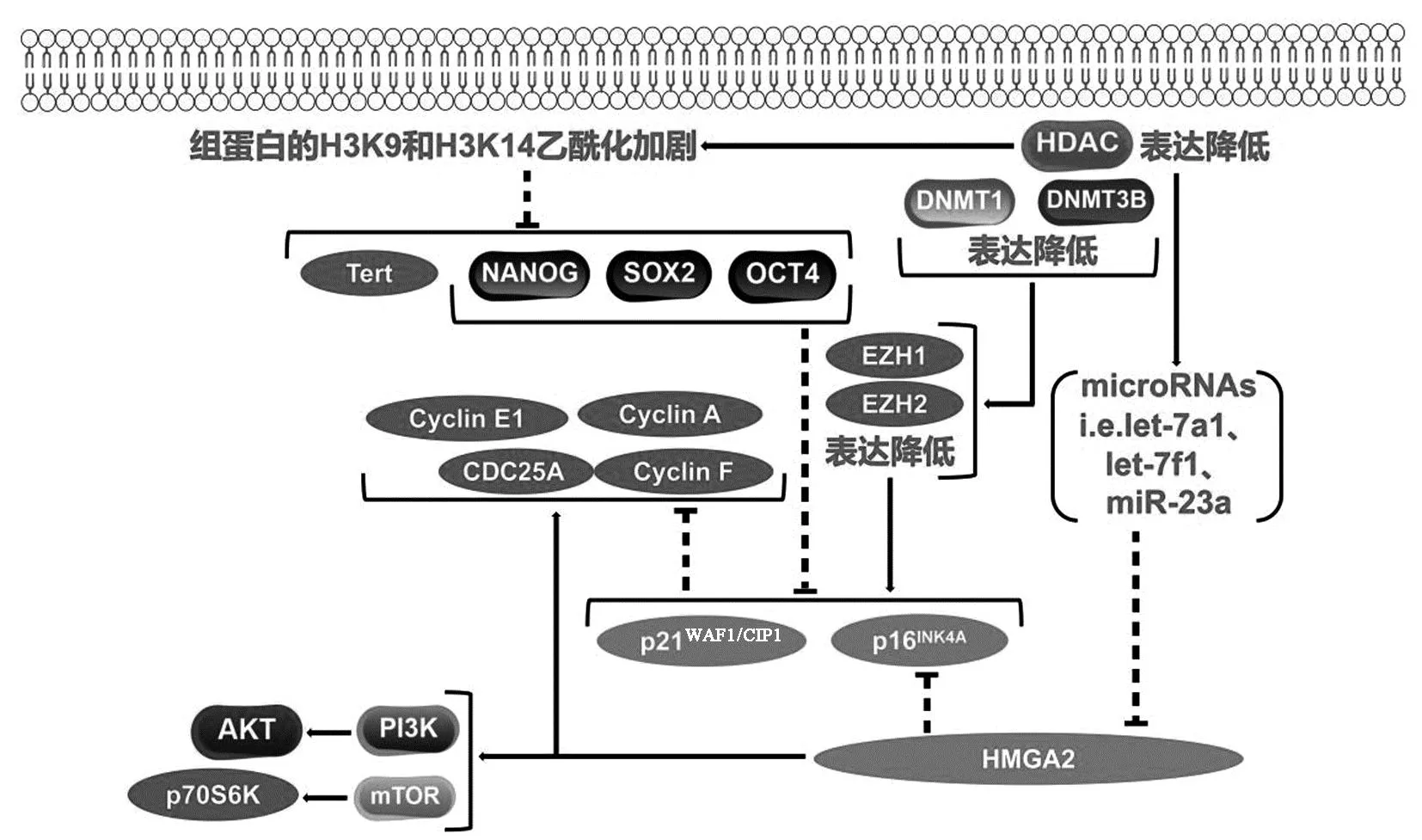
圖3 衰老BM-MSCs中的表觀遺傳修飾因子及下游靶基因表達變化Fig.3 Expression changes of epigenetic modification factors and downstream target genes in senescence BM-MSCs
1.2.2衰老過程中BM-MSCs的microRNAs及其下游靶基因表達變化:作為重要的表觀遺傳調控因子,眾多microRNAs在DDR、氧化應激和線粒體功能障礙等因素對BM-MSCs的衰老誘導中發揮重要作用。miR-21直接靶向抑制Sox-2表達,或通過降低Nanog和Oct4表達間接抑制Sox-2表達[20-21]。miR-200c和miR-155抑制線粒體活性并促進ROS生成[22-23]。miR-195通過抑制Tert、Sirt1、AKT和FOXO3的磷酸化表達,誘導端粒損傷并抑制自噬進而促進BM-MSCs衰老[24]。Epas1是重要的過氧化物酶體選擇性自噬調節蛋白,也是miR-142的靶基因。衰老BM-MSCs中,miR-142表達顯著上調并靶向抑制Epas1活性表達,導致ROS升高和受損過氧化物酶體積累[25]。隨著年齡增長,BM-MSCs中的miR-31a-5p表達上調,抑制E2F2和SATB2轉錄促進SAHF結構形成,進而誘導BM-MSCs衰老[20-21]。miR-204和miR-320調控IL-6和MMP-3等SASP因子分泌,并靶向抑制Runx2等成骨分化基因表達[26-27]。miR-34a介導p53對Nampt-NAD+-Sirt1信號通路的抑制,通過代謝障礙誘導MSCs衰老[28]。miR29b-1-5p的靶基因SDF-1可增強BM-MSCs的募集、增殖分化,以及在氧化應激中的自噬,衰老骨髓微環境激活miR29b-1-5p,降低SDF-1表達,誘導BM-BMSCs衰老[20-21]。
1.3 衰老BM-MSCs中發生細胞骨架重組和遷移歸巢能力下降
衰老BM-MSCs的肌動蛋白應力纖維增加而微管結合蛋白數量減少,使細胞內的力學結構變得更加均勻,細胞核更加收縮、細胞質中積累更多的顆粒和包涵體,導致細胞形態由典型的收縮紡錘形轉變為擴張扁平且不均勻的顆粒煎蛋狀,與塑料表面的粘附能力、集落形成能力和運動遷移能力均顯著降低[29]。
1.4 衰老BM-MSCs呈現SASP表型
衰老BM-MSCs呈現獨特的SASP表型,釋放多種促炎因子以自分泌的方式穩定自身衰老,并通過旁分泌誘導鄰近BM-MSCs等成骨譜系細胞中ROS生成增加和促進破骨細胞生成,加劇BM-MSCs的免疫調節和生態位支持功能受損,并增強全身水平的炎癥反應過度激活免疫系統,被認為是誘發持續性慢性炎癥和年齡相關疾病的重要原因[30]。隨著年齡增長,全身和骨髓微環境中出現顯著的IL-6等促炎因子水平升高和IL-10等抗炎因子水平降低,并與BM-MSCs的TGF-β、NF-κB、JAK-STAT3和MAPK等信號通路激活和免疫調節功能降低密切相關,是骨質疏松癥與類風濕性關節炎等發生和維持的重要因素。IL-4和IL-13可觸發BM-MSCs中的miR-142表達,通過氧化應激誘導BM-MSCs衰老[29]。TGF-β的分泌水平在衰老BM-MSCs中增加,并刺激各種細胞因子、炎癥介質和其他活性物質分泌,以及影響細胞外基質的合成和降解,抑制TGF-β受體可促進體外培養BM-MSCs的擴增和維持未分化狀態。NF-κB信號通路在BM-MSCs衰老過程中激活并促進TNF-α和IL-1β等促炎因子分泌,而TNF-α和IL-1β又可激活NF-κB信號通路,形成正反饋加劇BM-MSCs衰老[29,31]。
2 衰老導致BM-MSCs的細胞功能降低
2.1 衰老BM-MSCs喪失成骨與脂肪分化平衡
作為成骨細胞和骨髓脂肪細胞的共同祖細胞,BM-MSCs的成骨與脂肪分化平衡對維持骨脂平衡、骨穩態和重建骨代謝至關重要。隨著年齡增長,氧化應激和DNA損傷積累在BM-MSCs中誘發其分化向脂肪方向傾斜,Runx2/CBFα1、Osterix、Alp和Ocn等成骨分化基因的表達和鈣沉積形成顯著降低,而C/EBP、PPARγ和Lpl等脂肪分化基因的表達和脂滴形成顯著增加[5,32-33]。衰老BM-MSCs的自我更新和成骨分化逐漸降低甚至喪失,而成脂分化明顯增加,造成骨組織穩態紊亂和再生受損;同時,衰老BM-MSCs的SASP因子促進破骨細胞生成和成骨細胞減少,損傷成骨細胞的骨形成和破骨細胞的骨吸收之間的平衡,加劇骨形成損傷并引發骨丟失和骨質疏松癥等病變[34-35]。
2.2 衰老導致BM-MSCs的免疫調節功能降低
衰老對BM-MSCs在誘導巨噬細胞極化、抑制淋巴細胞增殖分化、應對促炎信號的遷移能力、以及平衡造血干細胞(hematopoietic stem cells,HSCs)的淋巴系與髓系分化等功能均產生重要影響,造成BM-MSCs的免疫調節功能受損和促炎因子表達升高,在自身免疫性疾病、炎癥性疾病和退行性疾病的病理進程中發揮重要推動作用[36-37]。MSCs的免疫調節活性并不總是抑制性的,而是在本質上取決于所暴露微環境中不同的微環境因子[37]。MSCs通過分泌免疫抑制因子或直接的細胞-細胞間相互作用誘導并維持巨噬細胞的抗炎表型,而這兩種過程均依賴于微環境中存在短暫但濃度很高的促炎信號刺激。隨著年齡增長,骨髓組織中逐漸呈現出長期存在而維持在低水平的IFN-γ等促炎因子和相當水平的抗炎因子,構成慢性炎癥微環境,通過TLR4調控BM-MSCs向免疫刺激表型轉換,產生更多的促炎因子而阻礙炎癥解決[38]。復制性衰老的BM-MSCs中FAS-L表達顯著降低,誘導T細胞凋亡和抑制其增殖的能力減弱;同時,抑制T細胞分泌IFN-γ與TNF-α等促炎因子和促進其分泌IL-10等抗炎因子的功能也降低。衰老BM-MSCs呈現促炎的SASP表型,高表達IL-1、IL-6、IL-8、TNF-α、MIF、MCP1和GM-CSF等促炎因子并招募炎癥細胞;而IL-2和IL-10等抑炎因子的分泌水平則顯著降低,導致微環境中促炎表型巨噬細胞和T細胞的數量和遷移增加,Th1細胞和Th17細胞擴增,并過度產生TNF-α和IL-17等炎性因子[39-40]。衰老BM-MSCs分泌組中眾多的炎性因子在系統水平上加劇炎癥反應,過度激活免疫系統、誘發免疫衰老及促進癌細胞增殖和遷移[37]。
2.3 衰老BM-MSCs的干細胞生態位支持功能下降并引起骨髓造血重建惡化
隨年齡增長而頻繁出現的突變可誘發骨髓微環境老化和免疫細胞衰老,并逐漸導致機體的炎癥狀態;同時,骨髓中的HSCs也經歷分化與自我更新的下降[41]。BM-MSCs的生態位支持功能對維持骨髓微環境和造血功能具有重要意義。然而,衰老導致BM-MSCs失去分化平衡,骨髓生態位和造血支持功能下降,骨髓中出現以犧牲骨發育為代價的脂肪積累,反過來又抑制骨髓的成骨再生和造血[6,10,17]。分化的脂肪細胞可中斷淋巴細胞生成,抑制HSCs的淋巴系分化而促進髓系分化,造成骨髓的髓系偏倚和造血功能失調,增加骨髓生成的同時加劇骨髓造血重建惡化。同時,衰老BM-MSCs與炎癥發展一致的SASP表型導致其與HSCs之間的通訊異常造成無效造血,誘發骨髓增生異常綜合征;并促進微環境中ASXL1等基因突變的反復積累,引發造血系統基因組不穩定,驅動白血病等惡性腫瘤發生[17,40-42](圖4)。骨髓增生異常綜合征、椎間盤退變和哈欽森吉爾福德綜合征等衰老相關疾病患者的髓核中均被發現有衰老BM-MSCs的積累[5-6]。
3 預防和逆轉BM-MSCs衰老的策略
3.1 小分子化合物和抑制劑
阿司匹林和維生素C等通過激活內源性端粒酶促進BM-MSCs再生增殖和成骨分化,但過度的端粒酶激活可誘發其惡性轉化。N-乙酰-L-半胱氨酸、異硫氰酸鹽和抗壞血酸等抗氧化劑,以及雷帕霉素等mTOR和MAPK信號通路抑制劑可改善氧化損傷恢復衰老MSCs功能[7,10-11,40]。
3.2 中藥活性成分
槲皮素通過調節Wnt-β-catenin、MAPK和PI3K-AKT信號通路抑制BM-MSCs衰老[42-43]。左歸丸通過抑制Wnt-β-catenin信號通路改善DNA損傷來延緩BM-MSCs衰老[44-45]。川芎嗪通過抑制NF-κB信號通路延緩BM-MSCs衰老,增強其自我更新和分化平衡并降低促炎因子分泌[42-43,46]。黃芪通過調節鈣磷代謝抑制BM-MSCs衰老促進其成骨分化[47]。
3.3 生長因子和激素
FGF-2和PDGF等生長因子顯著延緩MSCs衰老,促進其增殖和分化平衡[30]。MIF通過激活自噬恢復衰老BM-MSCs活力[48]。褪黑素通過上調Sirt1活性表達來抑制ROS的積累和p53、ERK與p38的活性表達,最終抑制BM-MSCs衰老[29]。
3.4 細胞基因工程
過表達Sirt3可通過降低ROS水平和抑制p16INK4A與p21WAF1/CIP1的活性表達實現對衰老MSCs的逆轉。過表達天冬氨酸β-羥化酶通過抑制GSK3β介導的Wnt信號通路延緩BM-MSCs衰老并調控其成骨分化。過表達Tert可恢復衰老BM-MSCs的正常核型和再生分化能力[30,49]。過表達SATB2顯著增強BM-MSCs的多能性轉錄因子表達和自噬,進而抑制其衰老[50]。敲除p16INK4A或沉默Rb可減少DNA損傷而抑制MSCs衰老,但沉默腫瘤抑制基因會破壞MSCs分化平衡并增加腫瘤發生風險[7-8]。
4 小結與展望
衰老損害BM-MSCs功能,并導致組織器官功能下降、個體衰老和相關疾病、以及腫瘤發生。衰老過程中,各種細胞特性發生相互影響的顯著改變,然而目前對衰老BM-MSCs各種特性變化之間分子關聯的研究尚鮮有報道。Nanog等多能性轉錄因子在BM-MSCs中呈現極低的表達水平,過表達這些多能性轉錄因子對衰老BM-MSCs的逆轉作用可能是作為某些核心調控因子的替代來實現的,而BM-MSCs隨衰老過程逐漸增強的異質性特性可成為通過單細胞測序技術挖掘這些核心調控基因的天然優勢。骨髓微環境中的代謝產物和調控因子對BM-MSCs的細胞功能產生重要影響;同時,衰老BM-MSCs的SASP表型也影響骨髓微環境的分子信號組成。目前,對骨髓微環境惡化與BM-MSCs衰老之間的分子關系和由之引發骨組織退行性疾病的分子細節尚缺乏深入探討。骨髓微環境信號因子對BM-MSCs分化平衡和其與免疫細胞及造血干細胞互作的影響主要是通過改變其染色質結構實現的,深入探索BM-MSCs衰老過程中染色質結構與分泌基因表達譜變化之間相互關聯的分子細節有助于更完整和詳細地揭示骨髓微環境惡化與BM-MSCs衰老誘發之間的相互關系。目前,更多的干預措施需要被開發出來以提高BM-MSCs臨床應用的有效性和安全性。中藥活性成分和小分子化合物與基因工程技術相結合將在這一領域中發揮巨大作用,中藥的多靶點特性可在逆轉BM-MSCs衰老分子通路的探明中發揮優勢,而小分子化合物則以其低廉方便組合靈活的優勢成為一些珍貴藥材的替代,最后通過基因工程技術可獲得穩定遺傳的衰老抑制細胞系。
猜你喜歡
中華詩詞(2022年6期)2022-12-31 06:41:24
鴨綠江(2021年35期)2021-04-19 12:24:18
考試與評價·高一版(2020年6期)2020-11-02 02:45:24
中國生殖健康(2019年3期)2019-02-01 06:12:26
中國科技論壇(2017年7期)2017-07-25 08:49:53
媽媽寶寶(2017年2期)2017-02-21 01:21:24
國際漢語學報(2016年1期)2017-01-20 08:21:20
鑿巖機械氣動工具(2016年3期)2016-03-01 04:00:25
海軍航空大學學報(2015年3期)2015-11-11 17:20:00
中國中醫藥現代遠程教育(2014年22期)2014-03-01 04:32:55
