藏藥阿如久巴丸質量標準研究
2024-02-23 13:26:12次仁旺姆楊麗瓊達娃卓瑪
中國民族民間醫藥 2024年2期
謝 平 次仁旺姆 楊麗瓊 達娃卓瑪
西藏自治區食品藥品檢驗研究院/國家藥品監督管理局中藥(藏藥)質量控制重點實驗室,西藏 拉薩 850000
阿如久巴丸(散)系藏族驗方,目前生產廠家有金哈達藏藥廠、西藏門孜康制劑中心、神猴藏藥廠等。阿如久巴丸即十味訶子丸,但為尊重傳統用藥習慣,在藥品注冊中由訶子、藏茜草、紅花、刀豆、豆蔻、山礬葉、紫草茸、松蒂等十一味藥味加工制成丸劑水丸,呈棕灰色至棕褐色,氣芳香,味微酸、苦。具有清腎熱、止痛、利尿等作用。臨床上用于腎炎、泌尿生殖系統炎癥及結石引起的尿頻、尿急、尿痛、下腹部疼痛、腰膝酸痛、拖足跛行、下肢麻木等[1-2]。
本研究通過顯微鏡觀察所收集到的制劑,能夠準確描述制劑粉末的顯微特征;采用薄層色譜法進行鑒別,對點樣量、展開劑進行考察,確定最佳色譜條件;對檢查項下的水分、灰分以及酸不溶性灰分等進行測定;并對浸出物含量進行了測定。本研究對具有阿如久巴丸生產批準文號的生產企業及制劑室進行樣品和原料藥材的收集,依次對收集的樣品展開上述項目的質量標準研究,研究過程中參考了《中國藥典》《中華人民共和國衛生部藥品標準(藏藥)》1995年版第一冊以及十味訶子丸(阿如久巴丸)質量標準研究的文獻報道[1,3,4]。擬定訶子、紅花和藏茜草的顯微鑒別;擬定訶子和紅花藥味的TLC鑒別。本研究對藏藥阿如久巴丸質量控制提供了技術支持。
1 儀器與試藥
1.1 儀器 正置光學顯微鏡(日本尼康,Nikon Eclipse E1000);電子天平(AL104型,瑞士METTLER-TOLEDO有限公司);紫外燈分析儀(ZF-I型,上海顧村電光儀器廠);薄層層析硅膠板:①硅膠G板(青島海洋化工有限公司);②硅膠G板(煙臺江友硅膠開發有限公司);③硅膠G板(青島海浪硅膠干燥劑公司)。電熱鼓風恒溫干燥箱、馬弗爐、可控調溫電爐、電熱恒溫水浴鍋等。
1.2 試藥 本實驗所使用的藥材來自5個廠家共5個批次的樣品,具體情況見表1。紅花對照藥材購自中國食品藥品檢定研究院(批號:110709-200803);訶子對照藥材購自中國食品藥品檢定研究院(批號:121015-201605)。二甲苯、中性樹膠、分析純鹽酸,超純水、甲醇、無水乙醇、碘化鉍鉀、濃硫酸等所用試劑均是國產分析純。

表1 阿如久巴丸企業信息及樣品批號
2 方法與結果
2.1 顯微鑒別 取5批阿如久巴丸樣品粉末及按處方自配阿如久巴丸粉末2g,過4號篩。將過篩后的粉末用解剖針刮出均勻涂在載玻片上,滴加稀甘油封片,蓋上蓋玻片后分別在40×、100×、400×顯微鏡下觀察。結果如圖1所示。

圖1 阿如久巴丸粉末鑒定特征圖
根據以上顯微結果,標準敘述為:取本品,置顯微鏡下觀察石細胞類方形、類多角形或呈纖維狀,直徑14~40 μm,長至130 μm,壁厚,孔溝細密(訶子)。有長管分泌細胞,含黃棕色至紅棕色分泌物;花粉粒類圓形、橢圓形或橄欖形,直徑約至60 μm,具3個萌發孔,外壁有齒狀突起(紅花)。非腺毛基部膨大,整體呈長圓錐狀;木纖維孔紋明顯;紅棕色內含物多散在(藏茜草)。
2.2 薄層色譜鑒別
2.2.1 訶子薄層色譜鑒別[2-5]稱取阿如久巴丸樣品粉末2 g,置具塞錐形瓶中,加乙酸乙酯50 mL,超聲處理20 min,濾過,濾液濃縮至1 mL,作為供試品溶液。按處方中各藥味的比例,自配不含訶子的藥材粉末作為陰性樣品,按照供試品溶液的制備方法,制成陰性對照溶液。取訶子對照藥材0.5 g,同供試品溶液制備方法制備對照藥材溶液。將上述制備的供試品,陰性對照以及對照藥材溶液用微量進樣器點樣于同一硅膠G薄層板,供試品溶液和陰性對照溶液各取10 μL,對照藥材溶液3 μL,展開劑用三氯甲烷-丙酮-甲酸(7∶2∶1),待其預飽和20 min后,展開,取出,晾干,噴以2%三氯化鐵乙醇溶液,105 ℃加熱至斑點顯色清晰。供試品色譜中,觀察到各批次供試品溶液與對照藥材溶液色譜相應的位置上顯相同藍色斑點,斑點清晰。3批供試品色譜中,在與對照藥材色譜相應的位置上,顯相同顏色的斑點。該鑒別方法對該藥中訶子的鑒別具有很好的適應性,分離度好,Rf值適中,重現性較好。結果如圖2所示。
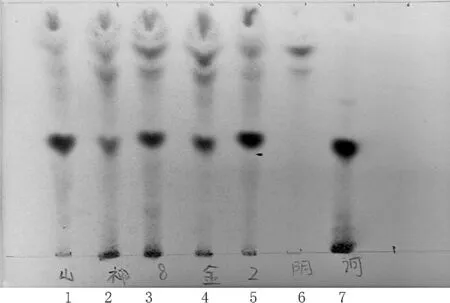
1~5.阿如久巴丸樣品;6.訶子陰性對照;7.訶子對照藥材圖2 訶子薄層色譜鑒別圖
本研究考察了3個展開系統,分別為三氯甲烷-乙醇-冰乙酸(6∶1∶0.1);三氯甲烷-乙酸乙酯-甲酸(5∶1∶0.2);三氯甲烷-丙酮-甲酸(7∶2∶1)。結果表明三氯甲烷-丙酮-甲酸(7∶2∶1)為展開系統時效果最好,斑點清晰,無拖尾,分離度良好,故以此為展開系統。同時本研究對點樣量也進行了考察,對照藥材溶液、供試品溶液點樣量考察了3 μL、5 μL、10 μL,結果表明對照藥材和供試品溶液分別為3 μL、10 μL時,斑點集中、清晰、不拖尾。故確定對照藥材溶液、供試品溶液點樣量分別為3 μL、10 μL。本研究對方法進行耐用性實驗考察,采用不同廠家的薄層板考察:①硅膠G板(青島海洋化工有限公司);②硅膠G板(煙臺);③硅膠G板(青島海浪硅膠干燥劑公司),展開劑為三氯甲烷-丙酮-甲酸(7∶2∶1),點樣量為供試品溶液、對照品溶液各10 μL、3 μL,顯色劑為2%三氯化鐵乙醇溶液,于日光下檢視。
結果表明,供試品在與對照藥材色譜相應的位置上,顯相同顏色的斑點。Rf值適中,分離度良好,斑點檢視清晰,分別采用不同廠家生產的硅膠G板按照上述方法進行耐用性實驗考察,結果在不同條件下該方法均能將樣品良好的分離,斑點檢視清晰,結果表明該方法耐用性良好。
2.2.2 紅花薄層色譜鑒別[1-4]稱取阿如久巴丸樣品粉末3 g,置于具塞錐形瓶中,加入80%丙酮5 mL,密塞,振搖15 min,靜置,取上清液作為供試品溶液。按處方中藥材的比例,自配不含紅花的藥材粉末作為陰性樣品,按照阿如久巴丸供試品溶液的制備方法,制成陰性對照溶液。取紅花對照藥材0.5 g,同供試品溶液制備方法制備對照藥材溶液。將上述制備的供試品,陰性對照以及對照藥材溶液用微量進樣器點樣于同一硅膠G薄層板,供試品溶液和陰性對照溶液,以及對照藥材溶液各取5 μL,展開劑為乙酸乙酯-甲酸-水-甲醇(7∶2∶3∶0.4),待預飽和20 min后,展開,取出,晾干。供試品色譜中,觀察到各批次供試品溶液與對照藥材溶液色譜相應的位置上顯相同顏色的斑點,斑點清晰。3批供試品色譜中,在與對照藥材色譜相應的位置上,顯相同顏色的斑點。該鑒別方法對該藥中紅花的鑒別具有很好的適應性,分離度好,Rf值適中,重現性較好。結果如圖3所示。

1.紅花對照藥材;2.紅花陰性對照;3~7.阿如久巴丸樣品圖3 阿如久巴丸(紅花)薄層鑒別圖(18 ℃,45%RH)
本研究考察了3個展開系統,并對其比例進行調整,分別為氯仿-丙酮-冰乙酸(6∶1∶0.1)、氯仿-乙酸乙酯-甲酸(5∶2∶0.2)、乙酸乙酯-甲酸-水-甲醇(7∶2∶3∶0.4),結果以乙酸乙酯-甲酸-水-甲醇(7∶2∶3∶0.4)為展開系統時效果最好,斑點清晰,無拖尾,分離度良好,故以此為展開條件。同時本研究對點樣量也進行了考察,對照品溶液、供試品溶液點樣量考察了3 μL、5 μL、10 μL,結果發現對照藥材溶液和供試品溶液為5 μL時,斑點集中、清晰、不拖尾。故確定對照藥材溶液、供試品溶液點樣量分別為5 μL。本研究對方法進行耐用性實驗考察,采用不同廠家的薄層板考察:①硅膠G板(青島海洋化工有限公司);②硅膠G板(煙臺);③硅膠G板(青島海浪硅膠干燥劑公司),展開劑為乙酸乙酯-甲酸-水-甲醇(7∶2∶3∶0.4),點樣量為供試品溶液、對照品溶液各5μL,于日光下檢視。
結果表明,供試品在與對照藥材色譜相應的位置上,顯相同的斑點。Rf值適中,分離度良好,斑點檢視清晰,分別采用不同廠家生產的硅膠G板按照上述方法進行耐用性實驗考察,結果在不同條件下該方法均能將樣品良好的分離,斑點檢視清晰,表明該方法耐用性良好。
2.3 阿如久巴丸水分含量、灰分及浸出物測定
2.3.1 水分測定 根據2020版《中國藥典》四部通則0832水分測定項下[6],選取第二法烘干法對阿如久巴丸進行水分含量測定。
實驗方法:差減法精密稱取阿如久巴丸藥材粉末3 g左右,平鋪于干燥至恒重的已編號稱量瓶中,厚度不超過5 mm,疏松阿如久巴丸藥材粉末不超過10 mm,打開瓶蓋在105 ℃下干燥5 h后將瓶蓋蓋好,移置干燥器中,冷卻至室溫,精密稱定重量。再在上述溫度干燥1 h,放冷,稱重,至連續兩次稱重的差異不超過5 mg為止。累計烘干6 h。根據公式計算阿如久巴丸樣品中含水量。
烘干法共測定5批阿如久巴丸,其中水分含量最高的批次為8.00%,最低為6.24%,平均值為6.99%。具體結果見表2。

表2 烘干法測定阿如久巴丸中水分含量結果表
實驗結果表明,阿如久巴丸不同來源樣品應用烘干法測定水分值相差最大在1.76%。按照2020版《中國藥典》四部通則0832水分測定法測定,5批樣品的水分均小于9.0%,符合規定。
2.3.2 灰分測定 根據《中國藥典》2020版第四部通則2302灰分測定法[6],對阿如久巴丸樣品進行總灰分及酸不溶性灰分的檢測。
實驗方法:差減法精密稱取5 g阿如久巴丸樣品于熾灼至恒重的已編號瓷坩堝中。在電爐上加熱30 min使樣品粉末碳化,待坩堝中無白煙冒出時表明樣品粉末已碳化完全。將坩堝放入馬弗爐中550 ℃下熾灼灰化2 h后取出,冷卻至150 ℃左右放入干燥器中冷卻至室溫,稱重,再放入馬弗爐中灰化1 h,取出,稱重。重復操作2次后達到恒重,累計熾灼4 h。根據公式計算阿如久巴丸中總灰分含量。
取上項所得的總灰分,在坩堝中加入10% 鹽酸溶液10 mL,蓋上蓋子,置水浴上加熱10 min,坩堝蓋用5 mL熱水沖洗,洗液并入坩堝中,用快速定量濾紙過濾,坩堝內的殘渣用熱水洗于濾紙上,并洗滌至洗液不顯氯化物反應為止。濾渣連同濾紙移置同一坩堝中,同時在另一坩堝中放入一張快速定量濾紙作為空白實驗。坩堝及樣品105 ℃下干燥,電爐上加熱至完全碳化后,移入馬弗爐中550 ℃熾灼至恒重。根據公式計算阿如久巴丸樣品中酸不溶性灰分的含量。
空白試驗及5批樣品結果見表3和表4,酸不溶性灰分空白試驗結果如表3所示;阿如久巴丸的總灰分含量最高為23.10%,最低為19.87%,平均值為21.43%。試驗結果見表4。

表3 酸不溶性灰分空白試驗結果 (n=2)
實驗結果表明阿如久巴丸的總灰分含量與酸不溶性灰分含量差異較大。推測阿如久巴丸中堿性無機成分含量較大的原因。各樣品總灰分平均為21.43%,上浮20%為25.72%。酸不溶性灰分平均含量為0.296%,上浮20%為0.355%。對阿如久巴丸的總灰分及酸不溶性灰分的檢測是保證阿如久巴丸用藥安全的一項重要指標,建議總灰分不得過26.0%,酸不溶性灰分不得過0.36%,一批不合格。

表4 阿如久巴丸的總灰分及酸不溶性灰分含量 (n=3)
2.2.3 浸出物測定 根據《中國藥典》2020年版第四部通則2201浸出物測定法[6],測定阿如久巴丸浸出物含量。
實驗方法:差減法精密稱取4 g阿如久巴丸樣品于250 mL錐形瓶中,精密加水100 mL,密塞,冷浸,前6 h內時時振搖,再靜置18 h,用干燥濾器迅速濾過,精密量取續濾液20 mL,置已干燥至恒重的蒸發皿中,在水浴上蒸干后,于105 ℃干燥3 h,置干燥器中冷卻30 min,迅速精密稱定重量。根據公式計算阿如久巴丸中浸出物的含量。
差減法精密稱取5份3 g上述同一批次阿如久巴丸樣品分別置于100 mL錐形瓶中,精密加水、30%乙醇、50%乙醇、70%乙醇及95%乙醇50 mL,密塞,稱定重量,再靜置1 h后,連接回流冷凝管,加熱至沸騰,并保持微沸1 h。放冷后,取下錐形瓶,密塞,再稱定重量,用所用溶劑補足減失的重量,搖勻,用干燥濾器濾過,精密量取續濾液25 mL,置已干燥至恒重的蒸發皿中,在水浴上蒸干后,于105 ℃干燥3 h,置干燥器中冷卻30 min,迅速精密稱定重量。根據公式計算阿如久巴丸中浸出物的含量。
取同一批次的阿如久巴丸加同一溶劑95%乙醇,分別考察了冷浸法與熱浸法,結果見表5。熱浸法所得的浸出物含量高于冷浸法所得。故進而選擇了熱浸法對5種溶劑進行了考察,所得結果見表6。根據表6結果,最終選定95%乙醇為浸出物提取溶劑,對5個批次進行浸出物含量考察。結果見表7。

表5 阿如久巴丸浸出物的冷浸法與熱浸法的考察 (n=3)

表6 阿如久巴丸浸出物提取溶劑的考察 (n=3)

表7 浸出物含量 (n=3)
實驗結果表明來自西藏雄巴拉曲制藥有限公司的阿如久巴丸的浸出物含量高,而來自西藏自治區藏醫院門孜康制劑室阿如巴丸浸出物含量低,推測原因可能與原藥材的產地、采收季節等因素有關,導致該批次的浸出物含量低。各批次浸出物含量平均值為33.35%。下浮20%為26.68%。對阿如久巴丸的浸出物含量檢測是保證該藥用藥安全的一項重要指標,建議浸出物含量不得少于27.0%,有一批浸出物含量不合格。
3 結論
本研究基于《中華人民共和國衛生部藥品標準(藏藥)》1995年版第一冊和中國藥典基礎上[3-4],通過顯微鑒別實驗,制定了阿如久巴丸中訶子、紅花、藏茜草顯微鑒別方法,通過薄層色譜實驗,制定了阿如久巴丸中訶子與紅花的薄層鑒別方法,完善了其質量標準,能有效幫助阿如久巴丸質量標準研究,為阿如久巴丸的質量標準建立奠定基礎。
