網(wǎng)絡(luò)化聚吡咯/聚氨酯復(fù)合材料的抗靜電性能研究
2024-02-28 07:18:44段書謙劉姝雅劉江慧成曉瓊張先群
中國塑料 2024年2期
關(guān)鍵詞:復(fù)合材料
段書謙,劉姝雅,劉江慧,成曉瓊,蒙 丹,張先群,陳 肖,付 海,2*
(1.貴州師范大學(xué)材料與建筑工程學(xué)院,貴陽 550025;2.貴州省教育廳輕質(zhì)材料工程研究中心,貴陽 550025)
0 前言
高分子材料普遍具有較高的電阻率、易產(chǎn)生靜電且靜電傳遞和釋放困難等特性,使其在生產(chǎn)和使用中存在安全隱患。目前靜電危害已成為高分子材料應(yīng)用中重要的問題之一[1]。為了解決這一問題,通過表面涂覆和添加導(dǎo)電劑提升高分子材料的電導(dǎo)性能,開發(fā)具有抗靜電性能的高分子復(fù)合材料。金屬類粉末與導(dǎo)電炭黑由于高的導(dǎo)電性被廣泛用于高分子抗靜電材料,但是由于金屬比重大、易腐、制備不便等缺點(diǎn),在高分子材料中的實(shí)際應(yīng)用受到局限;炭黑的微觀形態(tài)不易控制限制了更廣的應(yīng)用。導(dǎo)電高分子材料作為導(dǎo)電劑,因其大分子鏈與樹脂較好的相容性,且便于控制形貌,進(jìn)而有望發(fā)展成為一類新的導(dǎo)電劑品種[2]。
復(fù)合型高分子抗靜電材料的導(dǎo)電機(jī)理遵循著逾滲理論。因此導(dǎo)電劑在基體中的添加量直接影響著復(fù)合材料電導(dǎo)率的變化趨勢,這是由于在基體中導(dǎo)電劑足夠充分才能相互搭接形成導(dǎo)電網(wǎng)絡(luò)。導(dǎo)電網(wǎng)絡(luò)的構(gòu)建效率也會受其形貌、粒徑及分散狀態(tài)等因素的影響,故而催生了使用具有長徑比的納米線狀和二維片狀材料應(yīng)用于高分子抗靜電材料的深入研究。Wang[3]等將不同維度的碳系導(dǎo)電劑填充到異戊二烯橡膠中,具有高長徑比的一維碳納米管制備的復(fù)合材料具有最高的電導(dǎo)率。Yan[4]等將不同形貌特征的導(dǎo)電劑組合構(gòu)建導(dǎo)電網(wǎng)絡(luò)用于提高復(fù)合材料的電導(dǎo)性能。可見導(dǎo)電劑的形態(tài)對于導(dǎo)電網(wǎng)絡(luò)效能有著重要作用。當(dāng)高分子抗靜電材料在應(yīng)用環(huán)境中發(fā)生形變時,導(dǎo)電網(wǎng)絡(luò)搭接的穩(wěn)定性和健康程度影響著復(fù)合材料電導(dǎo)率的波動。不同維度和形態(tài)的導(dǎo)電劑所構(gòu)建的導(dǎo)電網(wǎng)絡(luò),在應(yīng)對材料形變時響應(yīng)結(jié)果會有所不同。本論文利用具有三維網(wǎng)絡(luò)形態(tài)的PPy 材料作為導(dǎo)電劑制備PU 復(fù)合材料,對比分析粒狀與網(wǎng)狀聚吡咯材料在復(fù)合材料中形成導(dǎo)電通路的效率。通過復(fù)合材料受力變形評價(jià)復(fù)合材料中導(dǎo)電網(wǎng)絡(luò)完整度與導(dǎo)電性能的關(guān)系,進(jìn)而探索導(dǎo)電劑的形態(tài)特征影響導(dǎo)電網(wǎng)絡(luò)穩(wěn)健性的因素,從而開發(fā)適應(yīng)實(shí)際場景的高分子抗靜電復(fù)合材料。
1 實(shí)驗(yàn)部分
1.1 主要原料
吡咯(Py),分析純,國藥化學(xué)集團(tuán)試劑有限公司;
十六烷基三甲基溴化銨(CTAB)、N,N-二甲基甲酰胺(DMF),分析純,國藥化學(xué)集團(tuán)試劑有限公司;
PU,K885,萬華化學(xué)集團(tuán)股份有限公司;
1.2 主要設(shè)備及儀器
掃描電子顯微鏡(SEM),ZEISS Sigma 300,德國ZEISS公司;
紅外光譜儀(FTIR),Tensor27,德國布魯克公司;
熱重分析儀(TG),STA449-F3,德國Netzsch公司;
動態(tài)熱機(jī)械分析儀(DMA),DMA8000,美國PerkinElmer公司;
高電阻測試儀,ZC36,上海雙旭電子有限公司;
全自動比表面積及孔隙度分析儀(BET),Tristar3020,麥克默瑞提克(上海)儀器有限公司;
1.3 樣品制備
將一定量Py 加入含CTAB 的HCl 水溶液(1 M)中,在上述混合溶液中加入過硫酸銨(APS)(Py 與APS 摩爾比為1/1),配方見表1;配置溶液需預(yù)冷至0~5 ℃,并持續(xù)攪拌反應(yīng)24 h;反應(yīng)結(jié)束后用去離子水和乙醇交替洗滌樣品3次以上,凍干至恒重得到黑色粉末;配方1、2和3制得樣品分別記為G-PPy、C-PPy和F-PPy);將制得粉末分散于DMF 中,在60 Hz 頻率下超聲30 min,后迅速將一定量的PU 粉末加入攪拌至完全溶解后向加入去離子水,然后將沉淀物烘至恒重,用平板硫化儀將在130 ℃、10 MPa 條件下熱壓10 min 制成片材待用,樣品名稱及配方見表2。
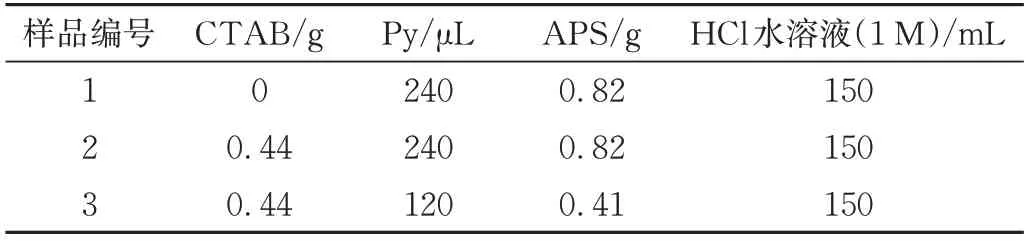
表1 PPy材料配方表Tab.1 Formula of PPy materials

表2 PPy/PU復(fù)合材料配方表Tab.2 Formula of PPy/PU composites
1.4 性能測試與結(jié)構(gòu)表征
紅外分析:將100 mg溴化鉀烘干,取1 mg樣品與之混合研磨,壓制成薄片;掃描范圍為400~4 000 cm-1;
形貌分析:用導(dǎo)電膠帶將試樣粘于樣品臺,噴金后進(jìn)行SEM表征,加速電壓為20 kV;
BET 分析:加熱溫度110 ℃、脫氣時間3 h、測定溫度-195.85 ℃、高純氬氣(≥99.999%);
熱重分析:取5~10 mg 樣品,在氮?dú)庀乱? ℃/min的速率從室溫升到800 ℃,考察其熱失重情況;
DMA 分析:在拉伸模式下進(jìn)行,實(shí)驗(yàn)溫度從-40~25 ℃,測試頻率為1 Hz,加熱速率為5 ℃/min;
導(dǎo)電性能分析:將試樣制成長4 cm,寬1 cm,厚2 mm 的樣條,使用高電阻測試儀的電極夾夾住樣條兩端測試;將試樣拉伸100%、200%、300%、400%、500%后保持2 cm 測試長度,測試應(yīng)變后樣條的兩端和中間處的寬度和厚度;利用電阻儀測得體積電阻(R,Ω),按照式(1)計(jì)算電阻率(σ,S/m):
式中l(wèi)——試樣長度,m
R——試樣電阻,Ω
S——試樣橫截面積,mm2
2 結(jié)果與討論
2.1 紅外分析
圖1中在3 431 cm-1處的寬峰為吡咯環(huán)的N—H 伸縮振動吸收峰,在1 529 cm-1處的吸收峰對應(yīng)吡咯環(huán)C=C 鍵的拉伸,而1 458 cm-1和1 169 cm-1處的吸收峰分別為C—N 鍵和C—H 鍵的平面變形,1 056 cm-1和884 cm-1處的吸收峰分別對應(yīng)于C—H 鍵的面內(nèi)彎曲振動和吡咯環(huán)C—H鍵的面外彎曲振動[5]。譜圖中3種Ppy材料出現(xiàn)的主要特征峰具有相同的峰位置,表明3種材料主要由Ppy 組成。C-PPy 和F-PPy 材料的譜圖中未出現(xiàn)CTAB 分子特征峰,表明CTAB 僅起到軟模板的作用誘導(dǎo)吡咯單體形成微結(jié)構(gòu)。

圖1 PPy材料的FTIR譜圖Fig.1 FTIR spectra of the PPy materials
2.2 形貌分析
從圖2(a)、(c)中觀察到明顯的線狀結(jié)構(gòu),線條細(xì)長且呈彎曲形態(tài)。圖2(b)、(d)進(jìn)一步顯示線條間相互連接形成網(wǎng)絡(luò)結(jié),C-PPy 與F-PPy 相比線徑稍粗。對圖2(b)、(d)中的聚吡咯納米線線徑分析,得到線徑尺寸分布圖[見圖2(a)、(c)嵌圖]。C-PPy 的線徑主要集中在30~50 nm 之間,而F-PPy的線徑主要集中在20~35 nm 之間。圖2(e)、(f)是在無模板劑條件制備的PPy,呈不規(guī)則顆粒狀,是由更小顆粒形成的團(tuán)聚體。團(tuán)聚體直徑約300~500 nm。PPy 納米線的形成主要依賴于CTAB 形成的蠕蟲狀膠束作為模板,膠束形態(tài)在表面活性劑濃度一定的條件下,其蠕蟲狀膠束內(nèi)部總體積處于平衡狀態(tài)。隨著吡咯單體增多,聚合形成的納米線線徑也會增加。因此C-PPy 相比F-PPy 納米線的線徑稍粗。G-PPy、C-PPy和F-PPy材料的粉末電導(dǎo)率分別為:0.099 5、0.001 2、0.001 4 S/m。其中CPPy 和F-PPy 的電導(dǎo)率值相近,而G-PPy 的電導(dǎo)率相比PPy 納米線幾乎高2 個數(shù)量級。這是因?yàn)轭w粒狀PPy 的粒徑較大,且顆粒聚集有利于降低接觸電阻提高電子傳輸速率。
2.3 BET分析
圖3 顯示了在77 K 下,P/P0=0.1~0.9 范圍內(nèi)的氮?dú)馕?脫附等溫線,分別對應(yīng)G-PPy、C-PPy 和FPPy 3 種聚吡咯材料。其中STP 表示在標(biāo)準(zhǔn)狀態(tài)下。3 條曲線都屬于典型的Ⅳ型:在較高的相對壓力區(qū)間,隨著相對壓力的增加,氮?dú)馕搅垦杆僭黾樱⒋嬖跍蟓h(huán)。G-PPy 和C-PPy 材料的比表面積為21.23 m2/g和161.14 m2/g,而F-PPy 的比表面積高達(dá)237.62 m2/g。這是由于其微觀結(jié)構(gòu)形態(tài)不同,所以比表面積差異較大。G-PPy 材料粒徑較大而且存在團(tuán)聚致使比表面積較低,而網(wǎng)絡(luò)化PPy 材料相較于顆粒團(tuán)聚體線徑尺度小,且存在多孔結(jié)構(gòu),比表面積較大。F-PPy 與CPPy相比線徑稍細(xì),因此比表面積也更大。
2.4 熱重分析
圖4(a)中3 種PPy 材料具有相似的熱重曲線,質(zhì)量損失率都達(dá)到了50%以上。其中100 ℃以下為試樣中水分的蒸發(fā),120~250 ℃為聚吡咯的初步分解,250~800 ℃為聚吡咯鏈的斷裂。為了進(jìn)一步探究3 種材料熱穩(wěn)定性的差異,對比質(zhì)量損失率為40%時的溫度,結(jié)果表明G-PPy 的熱分解溫度達(dá)到了688 ℃,而CPPy 和F-PPy 則分別為673 ℃和679 ℃,顆粒狀PPy 熱穩(wěn)定性稍優(yōu)于網(wǎng)絡(luò)狀PPy 的原因是,模板劑導(dǎo)致Py 聚合后存在低聚合度的PPy,在較低的溫度區(qū)間發(fā)生降解反應(yīng)[6]。

圖4 PU及PPy/PU復(fù)合材料的TG和DTG曲線Fig.4 TG and DTG curves of neat PU and PPy/PU composites
由圖4(b)在PPy/PU 復(fù)合材料中,PPy 的加入改善了PU 的熱穩(wěn)定性,其形貌的差異對熱穩(wěn)定性產(chǎn)生了不同的變化規(guī)律。根據(jù)表3 的數(shù)據(jù),PU 的起始分解溫度(t10)為321.05 ℃,半壽溫度(t50)為369.92 ℃。當(dāng)添加量為1%時,G-PPy、C-PPy 和F-PPy 的起始分解溫度分別提高了1.74、3.64、7.11 ℃,t50分別提高了4.68、4.68、14.55 ℃。而當(dāng)添加量為3%時,G-PPy、C-PPy和F-PPy 制備的復(fù)合材料的起始分解溫度分別提高了5.72、9.36、16.29 ℃,t50分別提高了12.3、23.39、24.43 ℃。由表可知,在導(dǎo)電劑添加量為1% ~3%時,網(wǎng)絡(luò)化PPy含量越多復(fù)合材料的熱穩(wěn)定性越強(qiáng)。
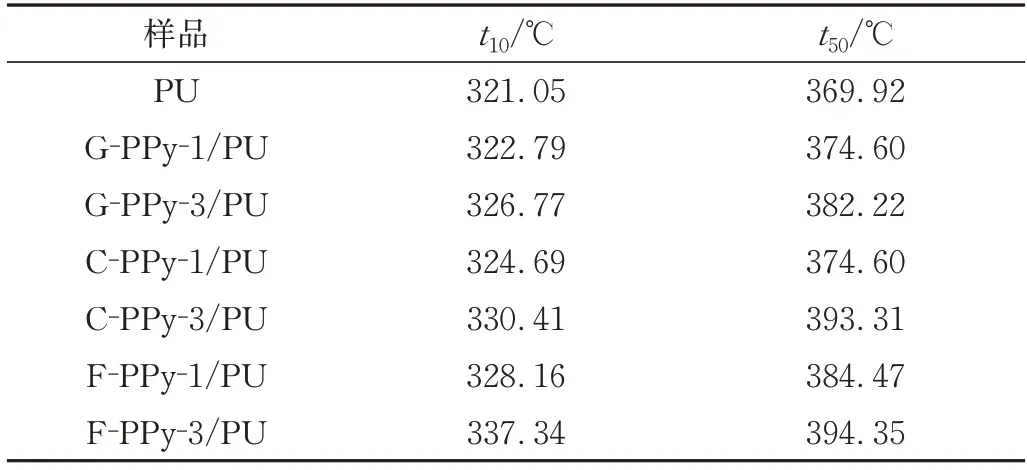
表3 PU及PPy/PU復(fù)合材料的各階段分解溫度Tab.3 Different stages of decomposition temperature of neat PU and PPy/PU composites
由圖4(c)分析得到PU 材料會有2 個質(zhì)量損失峰356 ℃和392 ℃,且在356 ℃時處質(zhì)量損失最大。PU 材料的分解過程主要分為2 個階段,即270~380 ℃和380~510 ℃這兩階段分別對應(yīng)軟鏈段和硬鏈段的降解[7]。添加PPy后通過曲線觀察發(fā)現(xiàn),當(dāng)填料形貌相同時,隨著PPy 含量的增加,復(fù)合材料的軟段分解峰面積逐漸減小,而硬段分解峰面積逐漸增大。當(dāng)填料含量相同時,線狀形貌材料制備的復(fù)合材料峰面積大于顆粒狀形貌,并且線徑越細(xì)趨勢越顯著。此外,熱分解過程表現(xiàn)出由2 個階段變?yōu)? 個高溫階段分解的趨勢[8]。這是因?yàn)榫W(wǎng)絡(luò)化PPy 相較于PPy 顆粒具有高的比表面積,填料與基體間的相界面面積越大,形成了具有更高熱穩(wěn)定性的界面過渡層。
2.5 DMA分析
圖5(a)~(b)為PU材料及PPy/PU復(fù)合材料儲能模量與溫度的關(guān)系曲線,對比分析得出,在整個溫度范圍內(nèi),復(fù)合材料的儲能模量均高于PU,并隨著溫度的升高而降低,并在玻璃化轉(zhuǎn)變溫度(Tg)附近出現(xiàn)平臺區(qū)。在導(dǎo)電劑含量為1%時,以網(wǎng)絡(luò)化PPy填充的復(fù)合材料的儲能模量遠(yuǎn)高于以PPy顆粒填充的復(fù)合材料,其中C-PPy-1/PU 和F-PPy-1/PU 的最大儲能模量分別為1 657 MPa和1 674 MPa,遠(yuǎn)高于G-PPy-1/PU的1 559 MPa。在導(dǎo)電劑含量為3%時,G-PPy-3/PU 的最大儲能模量為1 580 MPa,相較于G-PPy-1/PU略微提升,但仍低于同含量下以網(wǎng)絡(luò)化PPy為導(dǎo)電劑制備的復(fù)合材料,這是因?yàn)榫W(wǎng)絡(luò)化PPy導(dǎo)電劑相較于PPy顆粒比表面積大,與PU存在更大的界面面積,增大阻尼使得鏈段運(yùn)動困難[9]。

圖5 PPy/PU復(fù)合材料的動態(tài)熱力學(xué)分析Fig.5 Dynamic thermomechanical analysis of the PPy/PU composites
圖5(c)~(d)是PU 和PPy/PU 復(fù)合材料的損耗因子與溫度的關(guān)系曲線,通常以介質(zhì)損耗正切角(tanδ)的峰值溫度定義材料的Tg,從表4 復(fù)合材料Tg的變化可以看出,PPy 的引入使復(fù)合材料的Tg向高溫方向移動,這是因?yàn)镻Py 剛性粒子引入到高分子鏈段上,限制了高分子鏈段的運(yùn)動,鏈段在較高溫度時才能發(fā)生松弛。其中網(wǎng)絡(luò)化PPy 高的比表面積提供了更多連結(jié)點(diǎn),進(jìn)一步限制了鏈段的運(yùn)動能力,所以其制備復(fù)合材料的Tg變化更為顯著[10]。
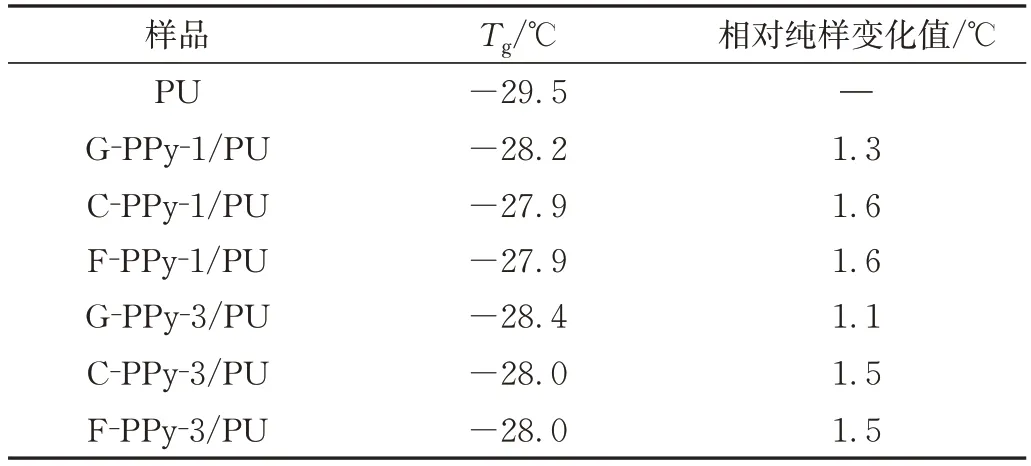
表4 PU及PPy/PU復(fù)合材料的TgTab.4 Tg of neat PU and PPy/PU composites
2.6 導(dǎo)電性能分析
圖6 為PPy/PU 復(fù)合材料在0~500%應(yīng)變條件下電導(dǎo)率的變化。對比圖中PU 和PPy/PU 復(fù)合材料在未形變下的電導(dǎo)率,聚吡咯作為導(dǎo)電劑的加入顯著提升了復(fù)合材料的電導(dǎo)率。在未形變下PU 的電導(dǎo)率為5.52×10-9S/m,而在加入1%的PPy 顆粒和網(wǎng)絡(luò)化PPy 導(dǎo)電劑后,PPy/PU 復(fù)合材料的電導(dǎo)率均達(dá)到了10-7S/m,提升了約3個數(shù)量級。當(dāng)復(fù)合材料受外力形變量逐漸增加時,電導(dǎo)率在100%應(yīng)變時均出現(xiàn)明顯下降,在大于100%應(yīng)變后,復(fù)合材料電導(dǎo)率下降幅度趨于平緩。在應(yīng)變?yōu)?00%、添加量為1%時,PPy 顆粒與網(wǎng)絡(luò)化PPy 所制備復(fù)合材料的電導(dǎo)率的下降幅度存在不同,G-PPy-1/PU 的電導(dǎo)率從10-7S/m 下降至3.9×10-10S/m,下降約3個數(shù)量級,而C-PPy-1/PU 和F-PPy-1/PU 的電導(dǎo)率從10-7S/m 分別下降至5.3×10-9S/m 和3.2×10-9S/m,低于G-PPy-1/PU 復(fù)合材料的下降率。在復(fù)合材料應(yīng)變100%~500%區(qū)間對比C-PPy-1/PU 和F-PPy-1/PU 復(fù)合材料與G-PPy-3/PU 復(fù)合材料電導(dǎo)率在形變下電導(dǎo)率下降趨勢發(fā)現(xiàn),GPPy-3/PU 的電導(dǎo)率從1.3×10-7S/m 下降至3.31×10-9S/m,與網(wǎng)絡(luò)化導(dǎo)電劑制備復(fù)合材料電導(dǎo)率的下降趨勢接近,顆粒的高濃度使得形成導(dǎo)電網(wǎng)絡(luò)結(jié)合點(diǎn)更多致使抵抗形變破壞的能力更強(qiáng)。綜合上述分析,在PPy導(dǎo)電劑添加1%時,PPy的網(wǎng)絡(luò)化形態(tài)更利于抵抗形變對導(dǎo)電網(wǎng)絡(luò)完整性的破壞作用(圖7)。
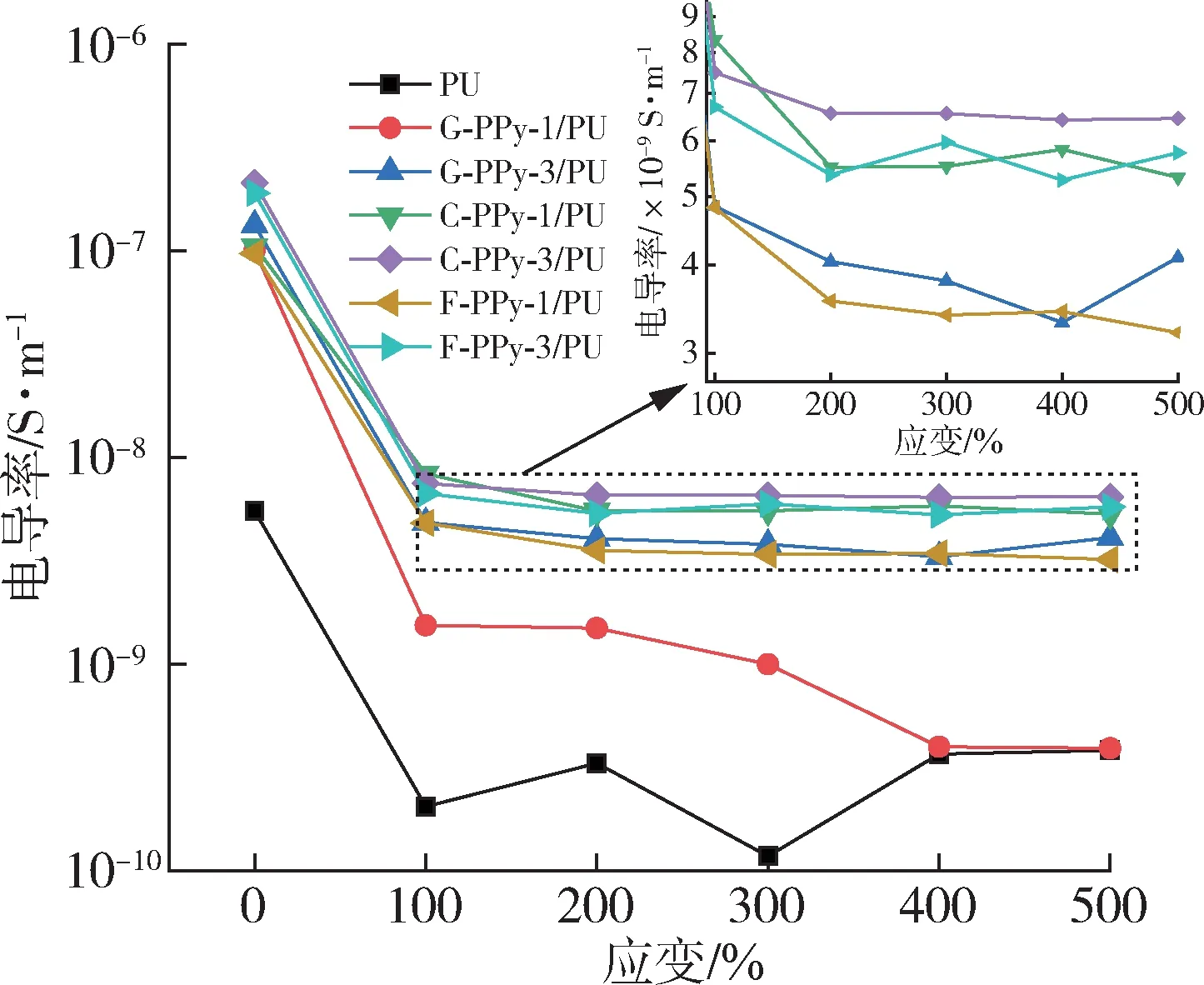
圖6 PU及PPy/PU復(fù)合材料的拉伸電導(dǎo)性能Fig.6 Tensile conductivity properties of neat PU and PPy/PU composites
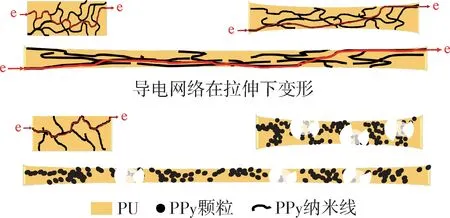
圖7 PPy/PU復(fù)合材料形變條件下的網(wǎng)絡(luò)變形Fig.7 Conductive network changes under deformation conditions of PPy/PU composites
C-PPy-1/PU、C-PPy-3/PU 復(fù)合材料和F-PPy-1/PU、F-PPy-3/PU 復(fù)合材料均為添加網(wǎng)絡(luò)化PPy 導(dǎo)電劑所制備的,其中C-PPy-1/PU、C-PPy-3/PU復(fù)合材料所用的C-PPy 材料相比F-PPy-1/PU、F-PPy-3/PU 復(fù)合材料所用的F-PPy具有更粗的線徑。因此C-PPy-1/PU、C-PPy-3/PU 復(fù)合材料中C-PPy 材料在超過導(dǎo)電劑的逾滲閾值后所形成的導(dǎo)電網(wǎng)絡(luò)具有更寬的導(dǎo)電路徑,即使形變后也更利于電子的快速傳遞,所以在導(dǎo)電劑含量相同下C-PPy/PU 復(fù)合材料的電導(dǎo)率略高于FPPy/PU復(fù)合材料。
3 結(jié)論
(1)利用膠束軟模板制得網(wǎng)絡(luò)化PPy材料,C-PPy材料和F-PPy材料的線徑分別在30~50 nm和20~35 nm之間,二者的微觀形態(tài)均呈現(xiàn)為納米線條構(gòu)成的網(wǎng)絡(luò)結(jié)構(gòu);
(2)對比PU材料,F(xiàn)-PPy-3/PU復(fù)合材料的起始分解溫度和半壽溫度分別提高了16.29 ℃和24.43 ℃。在PPy 導(dǎo)電劑添加量為1%時,網(wǎng)絡(luò)化PPy 比PPy 顆粒所制備復(fù)合材料的儲能模量高;
(3)PPy為導(dǎo)電劑制備PPy/PU 復(fù)合材料在500%應(yīng)變下,G-PPy-1/PU的電導(dǎo)率下降約3個數(shù)量級,而CPPy-1/PU和F-PPy-1/PU的電導(dǎo)率從10-7S/m分別下降至5.3×10-9S/m 和3.2×10-9S/m,下降率較低;這是由于PPy的網(wǎng)絡(luò)化形態(tài)利于抵御形變對導(dǎo)電通路完整性的不利影響,使得其電導(dǎo)率在變形時下降趨勢平穩(wěn)。
猜你喜歡
建材發(fā)展導(dǎo)向(2022年2期)2022-03-08 01:44:04
建材發(fā)展導(dǎo)向(2021年14期)2021-08-23 00:56:16
中國材料進(jìn)展(2019年10期)2019-12-07 05:32:14
纖維復(fù)合材料(2018年3期)2018-04-25 07:22:58
電子測試(2017年11期)2017-12-15 08:57:13
山東工業(yè)技術(shù)(2016年15期)2016-12-01 05:31:34
中國塑料(2015年6期)2015-11-13 03:02:54
中國塑料(2015年11期)2015-10-14 01:14:14
中國塑料(2015年8期)2015-10-14 01:10:41
應(yīng)用化工(2014年10期)2014-08-16 13:11:29
