細(xì)胞與基因治療臨床試驗(yàn)倫理審查實(shí)踐與分析
2024-02-29 06:15:44岳熠張雅麗薛貞雅陳碩劉利軍
中國醫(yī)藥生物技術(shù) 2024年1期
關(guān)鍵詞:研究
岳熠,張雅麗,薛貞雅,陳碩,劉利軍
細(xì)胞與基因治療(cell and gene therapy,CGT)是目前生物醫(yī)藥領(lǐng)域尤其是腫瘤、罕見病和遺傳病領(lǐng)域中極具發(fā)展前景的新一代精準(zhǔn)療法。細(xì)胞治療是指應(yīng)用人自體或異體來源的干/體細(xì)胞,經(jīng)分離、純化、培養(yǎng)、擴(kuò)增、活化等一系列體外操作后輸入(或植入)人體的治療方法;基因治療是指利用病毒或非病毒載體遞送作用將外源基因?qū)氚屑?xì)胞或組織,替代、補(bǔ)償、阻斷、修正特定基因,進(jìn)而實(shí)現(xiàn)從基因?qū)用嫔现委熌酥林斡膊〉母深A(yù)手段[1]。在現(xiàn)階段開展的CGT 臨床試驗(yàn)中,由于細(xì)胞來源、類型、操作等方面異質(zhì)性較大,治療原理和體內(nèi)作用等較傳統(tǒng)藥物更加復(fù)雜,CGT制劑的臨床療效、安全性風(fēng)險(xiǎn)、技術(shù)標(biāo)準(zhǔn)以及可能存在的倫理問題備受關(guān)注[2-3]。
倫理審查是 CGT 項(xiàng)目開展的重要“關(guān)卡”,符合CGT 技術(shù)特點(diǎn)、嚴(yán)謹(jǐn)規(guī)范的倫理審查與 CGT 臨床試驗(yàn)質(zhì)量直接相關(guān)[4]。本研究通過分析本院近五年 CGT 項(xiàng)目倫理初始審查情況,梳理歸納 CGT 項(xiàng)目審查重點(diǎn)難點(diǎn),探討構(gòu)建適應(yīng) CGT 新技術(shù)臨床研究活動(dòng)的倫理規(guī)范,為提高我國CGT 項(xiàng)目研究質(zhì)量,保障受試者安全與權(quán)益,推進(jìn)國內(nèi)CGT 臨床試驗(yàn)行業(yè)良性發(fā)展提供參考。
1 資料與方法
1.1 資料來源
2018 年 1 月 1 日 - 2022 年 12 月 31 日,本院CGT 臨床試驗(yàn)項(xiàng)目資料與倫理初始審查記錄。
1.2 數(shù)據(jù)收集
CGT 項(xiàng)目一般特征變量包括:項(xiàng)目類型[新藥注冊(cè)臨床試驗(yàn)(IND)、研究者發(fā)起的臨床試驗(yàn)(IIT)],制劑種類(干細(xì)胞制劑、CAR-T 細(xì)胞制劑、其他體細(xì)胞制劑、基因制劑),試驗(yàn)分期(I 期和 I/II 期、II 期、III 期)。審查決定分為“同意”“修正后同意”“修正后重審”和“不同意”4 類。審查決定為“修正后同意”“修正后重審”和“不同意”的項(xiàng)目均附有審查意見,參考《涉及人的生命科學(xué)和醫(yī)學(xué)研究倫理審查辦法》(2023)和《藥物臨床試驗(yàn)質(zhì)量管理規(guī)范》(2020)中有關(guān)研究方案、知情同意書的規(guī)定,將各項(xiàng)目審查意見中的倫理問題進(jìn)行歸納。
1.3 統(tǒng)計(jì)學(xué)處理
采用 SPSS 25.0 統(tǒng)計(jì)軟件進(jìn)行分析,分類變量以觀察值(構(gòu)成比)表示,對(duì)不同審查決定在不同項(xiàng)目特征下的發(fā)生頻次進(jìn)行χ2檢驗(yàn)或 Fisher 確切概率檢驗(yàn),設(shè)雙側(cè) α =0.05,P< 0.05 認(rèn)為存在統(tǒng)計(jì)學(xué)差異。
2 結(jié)果
2.1 初始審查基本情況
2018 - 2022 年,本院倫理委員會(huì)共受理、開展 CGT 臨床試驗(yàn)初始審查 74 項(xiàng),本單位作為組長單位/單中心開展的共 62 項(xiàng),初始審查平均一次性通過率為 36.50%(圖 1)。
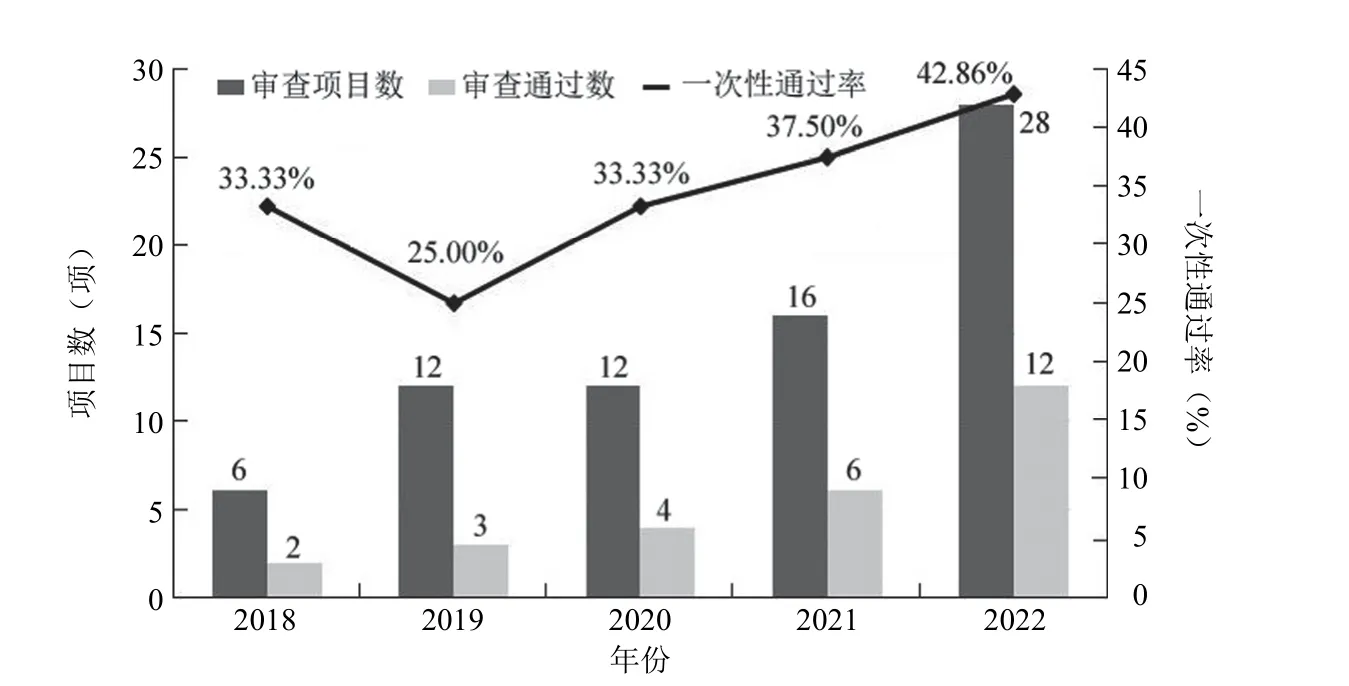
圖1 2018 - 2022 年 CGT 臨床試驗(yàn)倫理初始審查情況
2.2 項(xiàng)目基本特征與初始審查一次性通過率比較
項(xiàng)目類型構(gòu)成:IIT 研究 47 項(xiàng),IND 研究 27 項(xiàng)。制劑種類構(gòu)成:干細(xì)胞制劑 7 項(xiàng),CAR-T 制劑 45 項(xiàng),其他體細(xì)胞制劑 9 項(xiàng),基因治療制劑 13 項(xiàng)。試驗(yàn)分期構(gòu)成:I 期和 I/II 期 51 項(xiàng)、II 期 21 項(xiàng)、III 期 2 項(xiàng)。IIT 研究初始審查通過率低于 IND 研究,差異存在統(tǒng)計(jì)學(xué)意義(14.89% vs 74.07%,P< 0.0001)。不同制劑種類、試驗(yàn)分期的項(xiàng)目初始審查通過情況無統(tǒng)計(jì)學(xué)差異(表 1)。
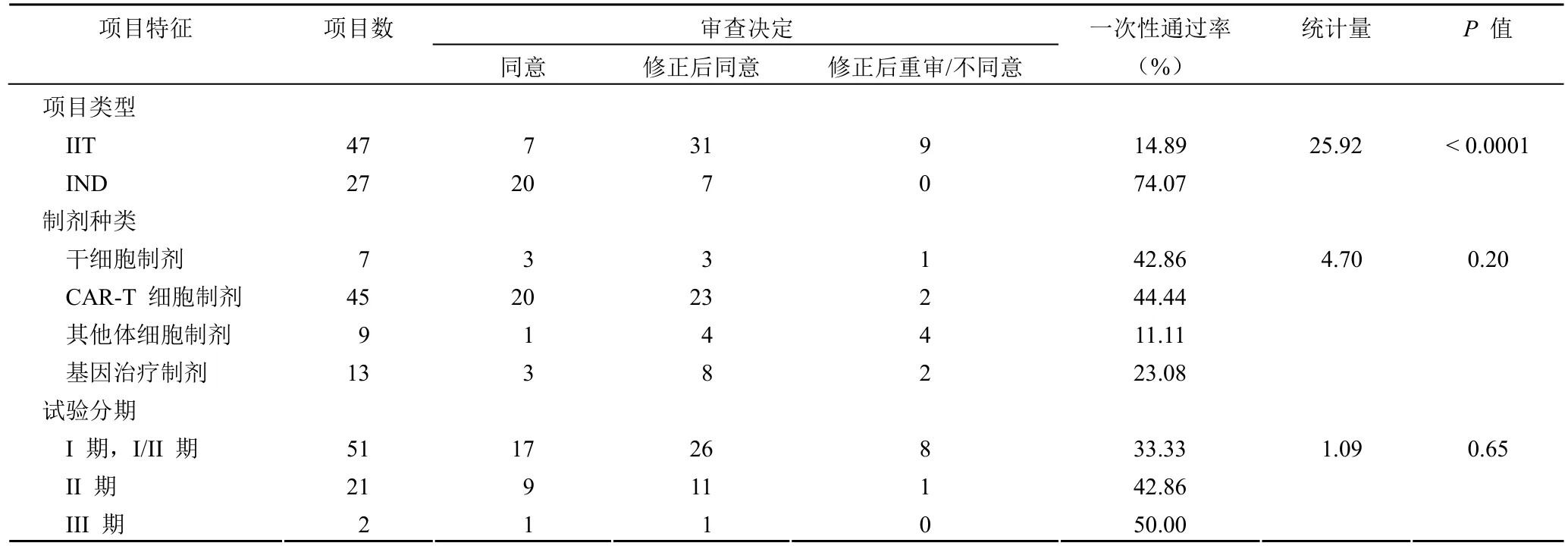
表1 2018 - 2022 年 CGT 項(xiàng)目初始審查一次性通過率比較
2.3 初始審查意見分析
本院 74 項(xiàng) CGT 臨床試驗(yàn)初始審查中,倫理委員會(huì)對(duì)其中 40 項(xiàng) IIT 研究和 7 項(xiàng) IND 研究未給予一次性通過且提出了修改意見。對(duì)意見匯總進(jìn)行倫理問題歸納。本院初始審查未通過的 CGT 項(xiàng)目中,倫理問題主要分布在研究基礎(chǔ)、知情同意書要素、研究設(shè)計(jì)和風(fēng)險(xiǎn)監(jiān)測與控制,詳見表 2。
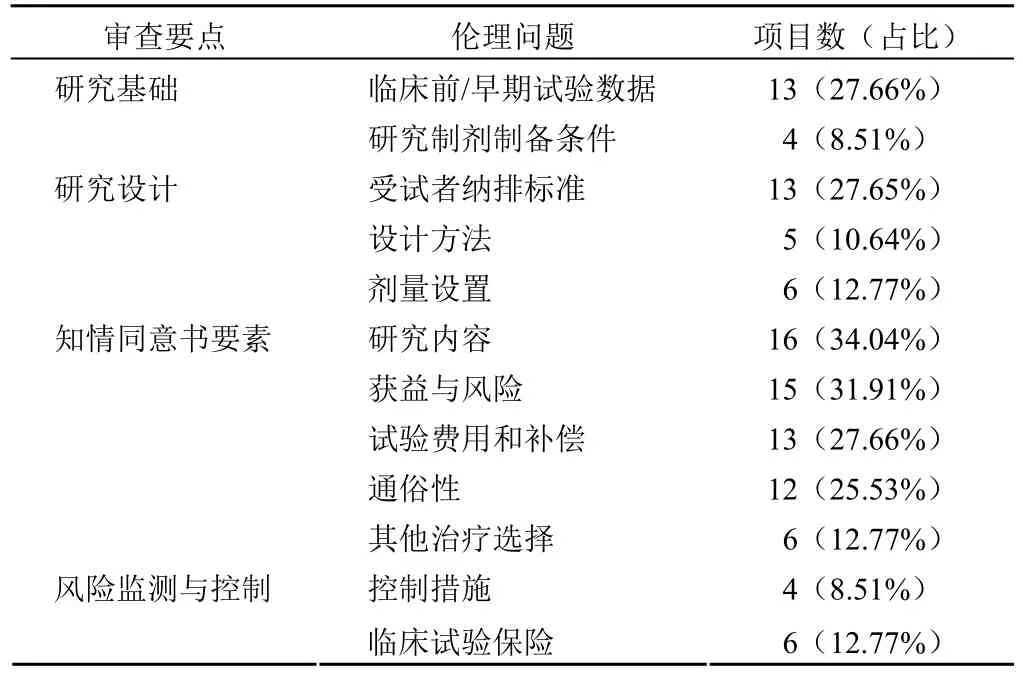
表2 CGT 臨床試驗(yàn)倫理問題分布
3 討論
我國 CGT 臨床試驗(yàn)實(shí)行“雙軌制”監(jiān)管模式,以新藥注冊(cè)為目的的臨床試驗(yàn)由國家食品藥品監(jiān)督管理局負(fù)責(zé)審批和監(jiān)管(IND-CGT);醫(yī)療機(jī)構(gòu)研究者發(fā)起的臨床試驗(yàn)由國家衛(wèi)生健康委員會(huì)采取備案制管理(IIT-CGT)。本院 CGT項(xiàng)目初始審查數(shù)據(jù)分析顯示 IIT-CGT 一次性通過率為14.89%,IND-CGT 一次性通過率為 74.07%,IIT-CGT 和IND-CGT 一次性通過率存在顯著差異,這提示 IIT-CGT 可能存在較多倫理問題。本研究進(jìn)一步對(duì)項(xiàng)目倫理審查意見進(jìn)行歸納總結(jié),發(fā)現(xiàn) CGT 的倫理問題主要發(fā)生在研究基礎(chǔ)、知情同意書要素、研究設(shè)計(jì)和風(fēng)險(xiǎn)監(jiān)測與控制方面,現(xiàn)結(jié)合本院倫理審查經(jīng)驗(yàn),以 CGT 常見倫理問題為切入點(diǎn),分析探討 CGT 項(xiàng)目倫理審查要點(diǎn)與對(duì)策。
3.1 CGT 項(xiàng)目倫理審查要點(diǎn)
3.1.1 研究基礎(chǔ) 臨床前/早期臨床試驗(yàn)數(shù)據(jù)是倫理委員會(huì)對(duì)藥物臨床試驗(yàn)項(xiàng)目預(yù)期風(fēng)險(xiǎn)獲益評(píng)估的重要依據(jù)。前期數(shù)據(jù)應(yīng)闡明藥物的作用機(jī)制、藥理毒理學(xué)特征,明確藥物在擬定適應(yīng)證中使用的生物學(xué)合理性、給藥劑量和給藥程序,提示藥物可能引發(fā)的不良反應(yīng)等。但 CGT 制劑有別于傳統(tǒng)小分子和大分子藥物,在臨床前/早期臨床試驗(yàn)階段,還應(yīng)充分考慮其種類多樣性、生物學(xué)復(fù)雜性和科學(xué)認(rèn)知局限性,在遵循創(chuàng)新藥物研發(fā)臨床前研究一般原則的同時(shí),結(jié)合每種CGT 制劑的特點(diǎn),采取“個(gè)體化”策略和基于風(fēng)險(xiǎn)的設(shè)計(jì)方法,具體問題具體分析,充分表征產(chǎn)品的藥理學(xué)、毒理學(xué)、藥代動(dòng)力學(xué)特征、量效關(guān)系特點(diǎn)和藥效活性[5]。本院部分CGT 項(xiàng)目臨床前數(shù)據(jù)不足的具體表現(xiàn)為:①動(dòng)物實(shí)驗(yàn)觀察期短,缺乏動(dòng)物模型接受研究制劑輸注后的生存信息;②研究制劑制備使用的慢病毒表達(dá)質(zhì)粒含有基因突變體,病毒載體介導(dǎo)的基因序列插入具有隨機(jī)性和不可預(yù)測性,缺乏評(píng)估該基因序列插入正常細(xì)胞基因組的風(fēng)險(xiǎn)評(píng)估信息;③研究制劑輸注劑量選擇和劑量爬坡終止標(biāo)準(zhǔn)依據(jù)不充分,缺乏殺傷人體正常細(xì)胞組織的實(shí)驗(yàn)數(shù)據(jù)。
在研究制劑的制備及質(zhì)量管理方面,2015 年 8 月國家衛(wèi)生健康委發(fā)布的《干細(xì)胞臨床研究管理辦法(試行)》中規(guī)定干細(xì)胞制劑的制備應(yīng)當(dāng)符合《藥品生產(chǎn)質(zhì)量管理規(guī)范》(GMP)的基本原則和相關(guān)要求,且干細(xì)胞制劑質(zhì)量管理應(yīng)遵照《干細(xì)胞制劑質(zhì)量控制及臨床前研究指導(dǎo)原則(試行)》。2022 年 10 月國家藥監(jiān)局發(fā)布的《細(xì)胞治療產(chǎn)品生產(chǎn)質(zhì)量管理指南》(試行)進(jìn)一步細(xì)化了細(xì)胞治療產(chǎn)品生產(chǎn)質(zhì)量管理的具體要求,2023 年 5 月發(fā)布的《體細(xì)胞臨床研究工作指引》(征求意見稿)中也涉及了體細(xì)胞制劑的供者篩查、人員設(shè)施、原輔料包材質(zhì)量、工藝質(zhì)控和追溯規(guī)程等相關(guān)內(nèi)容,提出應(yīng)不斷完善制劑質(zhì)量控制體系,提升體細(xì)胞臨床研究水平[6]。本院項(xiàng)目審查中有 4 項(xiàng) CGT 項(xiàng)目的制劑生產(chǎn)和質(zhì)控方面存在問題,例如:①缺少《藥品生產(chǎn)質(zhì)量管理規(guī)范》(GMP)符合性證明文件;②制劑制備的質(zhì)量管理體系有待進(jìn)一步完善,工藝驗(yàn)證及變更需更加規(guī)范。
3.1.2 研究設(shè)計(jì) 嚴(yán)密的臨床試驗(yàn)方案對(duì)保證 CGT 項(xiàng)目科學(xué)性、試驗(yàn)數(shù)據(jù)可靠性和保障受試者安全至關(guān)重要。在設(shè)計(jì)方法、受試者篩選、劑量設(shè)置等方面存在問題,可能影響對(duì)受試者安全性和真實(shí)療效的判斷。本院 CGT 項(xiàng)目研究設(shè)計(jì)共性問題主要表現(xiàn)在:①受試者目前存在有效治療方法,研究制劑輸注可能顯著增加安全性風(fēng)險(xiǎn);②受試者病情較重,無法等待細(xì)胞單采和制備時(shí)間;③首次人體試驗(yàn)未采用逐例入組、間隔給藥方式;④劑量探索方法和劑量爬坡終止標(biāo)準(zhǔn)不明確、試驗(yàn)劑量范圍相差 100 倍、劑量增幅過大。
3.1.3 知情同意書要素 受試者由于醫(yī)療專業(yè)知識(shí)限制,與研究者在 CGT 研究上信息不對(duì)稱,對(duì)于創(chuàng)新型生物醫(yī)藥制品,受試者可能會(huì)對(duì)其療效有過高預(yù)期。因此知情同意書是受試者了解擬參加 CGT 試驗(yàn)的重要文本和自主選擇是否參加 CGT 試驗(yàn)的主要依據(jù)。知情同意書必須采用與受試者文化程度相匹配的用語,充分、客觀告知受試者有關(guān)臨床試驗(yàn)的所有事宜,包括研究背景與目的、研究內(nèi)容與程序、獲益與風(fēng)險(xiǎn)、試驗(yàn)費(fèi)用與補(bǔ)(賠)償、其他治療選擇等。本研究顯示,CGT 項(xiàng)目知情同意書存在的倫理問題最多,例如:①研究者混淆使用“試驗(yàn)”“研究”“治療”“療法”等術(shù)語,未解釋試驗(yàn)與常規(guī)治療在受益和風(fēng)險(xiǎn)上的區(qū)別,可能使患者產(chǎn)生“治療性誤解”;②未能向受試者說明前期安全性和有效性數(shù)據(jù)、研究藥物劑量和需要配合的研究程序;③缺乏對(duì)可能發(fā)生的嚴(yán)重不良事件、遠(yuǎn)期風(fēng)險(xiǎn)及其臨床表現(xiàn)的描述;④未提供其他治療選擇的具體用藥及療效情況;⑤使用英文縮寫、醫(yī)學(xué)專業(yè)術(shù)語且未作口語化解釋,限制民事行為能力受試者版知情同意書缺失或用語不符合受試者認(rèn)知水平。
3.1.4 風(fēng)險(xiǎn)監(jiān)測與控制 患者既是 CGT 新技術(shù)研究的受益者,也是風(fēng)險(xiǎn)的承擔(dān)者。根據(jù)制劑特性制訂全面、可操作的風(fēng)險(xiǎn)監(jiān)測與處置預(yù)案對(duì)于試驗(yàn)風(fēng)險(xiǎn)控制和受試者保護(hù)具有重要意義。本研究發(fā)現(xiàn) CGT 項(xiàng)目風(fēng)險(xiǎn)監(jiān)測與處理方面的不足主要為:①遺漏某些發(fā)生概率低的致命性風(fēng)險(xiǎn),例如異體供者來源的制劑中存在免疫細(xì)胞,可能引起急性移植物抗宿主病、前期研究提示制劑存在重要非靶向器官大量聚積分布,若阻塞于肺部微血管可能引起急性肺栓塞;②項(xiàng)目未設(shè)置安全審查委員會(huì)評(píng)估安全性數(shù)據(jù);③保險(xiǎn)期限短于試驗(yàn)遲發(fā)安全性風(fēng)險(xiǎn)隨訪年限,保險(xiǎn)額度不能覆蓋研究相關(guān)不良事件的預(yù)期醫(yī)療費(fèi)用。
3.2 對(duì)策與建議
3.2.1 完善 CGT 制劑臨床前研究和質(zhì)量控制 CGT 制劑種類繁多,不同制劑的治療原理差別較大,體內(nèi)生物學(xué)行為如增殖、長期存活和持續(xù)作用也存在諸多不確定因素[7]。不同類型和用途的 CGT 制劑需要完成的臨床前研究要求可能不完全相同,國家衛(wèi)生健康委和國家藥監(jiān)局近年陸續(xù)發(fā)布的《干細(xì)胞制劑質(zhì)量控制及臨床前研究指導(dǎo)原則(試行)》《細(xì)胞治療產(chǎn)品研究與評(píng)價(jià)技術(shù)指導(dǎo)原則(試行)》《基因治療產(chǎn)品非臨床研究與評(píng)價(jià)技術(shù)指導(dǎo)原則(試行)》《基因修飾細(xì)胞治療產(chǎn)品非臨床研究技術(shù)指導(dǎo)原則(試行)》,在干細(xì)胞、體細(xì)胞和基因治療制劑毒性研究、安全性研究、異常免疫反應(yīng)研究、藥代動(dòng)力學(xué)研究、藥效學(xué)研究、成瘤性和致瘤性(致癌性)研究方面均做了詳細(xì)要求。在倫理審查實(shí)踐中,尤其對(duì)于 CGT 制劑首次人體試驗(yàn),倫理委員會(huì)在項(xiàng)目審查中對(duì)劑量設(shè)置依據(jù)和風(fēng)險(xiǎn)評(píng)估完全依賴于臨床前研究數(shù)據(jù)。在前期數(shù)據(jù)評(píng)估時(shí),倫理委員會(huì)主要關(guān)注的風(fēng)險(xiǎn)包括基因整合突變、致瘤性、生殖傳遞、在(脫)靶風(fēng)險(xiǎn)和免疫原性及其不良影響等。建議 CGT 項(xiàng)目申辦方或研究者充分開展非臨床研究,加強(qiáng)前期數(shù)據(jù)的完整度和可靠性,包括采用與預(yù)期人體反應(yīng)接近或相似的動(dòng)物模型或疾病細(xì)胞系深入探索劑量范圍和潛在毒性機(jī)制,完善相關(guān)毒性信息;對(duì)基因修飾的制劑進(jìn)行多批次測序,分析基因插入整合位點(diǎn),評(píng)價(jià)插入后基因突變風(fēng)險(xiǎn);關(guān)注動(dòng)物模型中制劑在體內(nèi)的植入、分布、分化、存續(xù)情況及與安全性有效性的關(guān)系等。當(dāng) CGT 制劑的生物學(xué)作用與實(shí)驗(yàn)系統(tǒng)、動(dòng)物種屬高度相關(guān),疾病模型構(gòu)建和動(dòng)物實(shí)驗(yàn)結(jié)論外推至人體存在局限性等特殊情況下,也可只完成概念性驗(yàn)證研究[6]。
CGT 制劑與傳統(tǒng)化藥相比,取材來源多樣、制備工藝復(fù)雜,制劑生產(chǎn)與臨床需求結(jié)合更為緊密,生產(chǎn)過程中發(fā)生污染、混淆、差錯(cuò)等風(fēng)險(xiǎn)的可能性更高[8]。在國內(nèi)“雙軌制”政策背景下,對(duì)于按照藥品研發(fā)和申報(bào)的 CGT 制劑的生產(chǎn)條件應(yīng)符合 GMP 的基本原則和相關(guān)要求;對(duì)于由醫(yī)療機(jī)構(gòu)研究者發(fā)起的、非藥品注冊(cè)為目的的 CGT 臨床研究,《干細(xì)胞臨床研究管理辦法(試行)》中規(guī)定干細(xì)胞制劑應(yīng)符合GMP 和《干細(xì)胞制劑質(zhì)量控制及臨床前研究指導(dǎo)原則(試行)》的要求,《國家醫(yī)療衛(wèi)生機(jī)構(gòu)開展研究者發(fā)起的臨床研究管理辦法(試行)》中規(guī)定非產(chǎn)品研制的體細(xì)胞臨床研究參照《干細(xì)胞臨床研究管理辦法(試行)》管理。另外,為進(jìn)一步加強(qiáng)體細(xì)胞臨床研究管理,2023 年中國醫(yī)藥生物技術(shù)協(xié)會(huì)發(fā)布的《體細(xì)胞臨床研究工作指引(試行)》中也新增了體細(xì)胞制劑應(yīng)達(dá)到的技術(shù)要求和質(zhì)控標(biāo)準(zhǔn)。因此從供者材料采集到制劑回輸全過程的標(biāo)準(zhǔn)操作程序、技術(shù)要求和產(chǎn)品質(zhì)控體系也是倫理委員會(huì)的審查重點(diǎn)之一。建議 CGT 制備機(jī)構(gòu)設(shè)置獨(dú)立的質(zhì)量管理部門,配備足夠數(shù)量的專業(yè)操作人員和管理人員并定期組織培訓(xùn)考核,針對(duì)具體研究制劑特性建立完整的生產(chǎn)質(zhì)量管理與控制體系,覆蓋制劑生產(chǎn)環(huán)境、設(shè)施設(shè)備、原輔料管理、制備流程、質(zhì)控標(biāo)準(zhǔn)、成品放行標(biāo)準(zhǔn)、污染防控與應(yīng)急預(yù)案等內(nèi)容。對(duì)于制劑生產(chǎn)工藝和質(zhì)控體系尚在驗(yàn)證優(yōu)化過程中的機(jī)構(gòu),至少應(yīng)保證工藝路線清晰、無外源因子污染和已知風(fēng)險(xiǎn)因素得到良好控制等要求,必要時(shí)可請(qǐng)上級(jí)管理部門、第三方機(jī)構(gòu)在項(xiàng)目開展前進(jìn)行生產(chǎn)制備平臺(tái)檢查評(píng)估[6]。
3.2.2 圍繞 CGT 特性制訂研究方案 由于 CGT 制劑具有特殊的生物學(xué)特性,CGT 項(xiàng)目需采用不同于傳統(tǒng)藥物的臨床試驗(yàn)設(shè)計(jì)策略,根據(jù)不同 CGT 制劑的作用特點(diǎn)及其機(jī)制,選擇合適的設(shè)計(jì)方法、研究人群和試驗(yàn)劑量[9]。選擇研究人群時(shí),應(yīng)重點(diǎn)考慮 CGT 作用機(jī)制與疾病診斷標(biāo)準(zhǔn)、分期特征、進(jìn)展速度和嚴(yán)重程度的適配性,以此預(yù)判受試者預(yù)期獲益和潛在風(fēng)險(xiǎn),從而制訂合理的納排標(biāo)準(zhǔn)。對(duì)于輸注制劑后需要較長時(shí)間隨訪才能顯現(xiàn)療效;自體原材料采集制備條件要求較高,晚期患者不能達(dá)到標(biāo)準(zhǔn)或不能耐受相關(guān)操作;極有可能早期治愈某種難治性疾病或極大程度延長患者生存期、改善患者生存質(zhì)量的項(xiàng)目,可考慮選擇病情較輕、分期較早、從試驗(yàn)中獲益可能性較大的患者,為患者爭取緩解癥狀、疾病康復(fù)的唯一機(jī)會(huì)[10]。確定研究劑量時(shí),應(yīng)基于合適的動(dòng)物模型、現(xiàn)有臨床經(jīng)驗(yàn)和目標(biāo)受試者風(fēng)險(xiǎn)承受能力估計(jì)起始劑量、劑量遞增范圍、劑量爬坡終止標(biāo)準(zhǔn)。設(shè)計(jì)研究方案時(shí),應(yīng)詳細(xì)規(guī)定最佳有效劑量的探索方法、輸注方式、間隔和頻率、既往用藥洗脫期和設(shè)定依據(jù)。為避免多個(gè)受試者同時(shí)暴露于非預(yù)期風(fēng)險(xiǎn),CGT 首次人體試驗(yàn)須采用逐例入組方式,上一例受試者的安全性和體內(nèi)制劑存活-作用時(shí)間結(jié)果評(píng)估通過后再入組下一例。
3.2.3 以患者為中心做好知情同意 醫(yī)療實(shí)踐的倫理核心為“個(gè)體患者受益”,而現(xiàn)有 CGT 項(xiàng)目的本質(zhì)是以當(dāng)前受試者為研究對(duì)象,獲得研究制劑可普遍化的有效性、安全性數(shù)據(jù),使未來患者獲益為目的的科學(xué)研究,必然存在科學(xué)知識(shí)積累的社會(huì)獲益和受試者個(gè)體獲益之間的沖突[11]。研究者作為 CGT 項(xiàng)目的主要實(shí)施人員,其倫理規(guī)范意識(shí)直接影響受試者權(quán)益保護(hù)機(jī)制的執(zhí)行[12]。研究者應(yīng)在入組前與受試者就知情同意書內(nèi)容展開深入討論,采用通俗易懂的語言向受試者正確解釋試驗(yàn)?zāi)康摹⒀芯績?nèi)容與程序、可能的獲益與潛在風(fēng)險(xiǎn),其他治療選擇及預(yù)期療效、試驗(yàn)相關(guān)損害及處理措施,避免誘導(dǎo)、欺騙與脅迫行為,確保受試者知情的信息真實(shí)可靠。在履行知情同意程序中,最重要的環(huán)節(jié)是幫助受試者認(rèn)識(shí)到參與試驗(yàn)可能不會(huì)帶來疾病緩解或治愈,也面臨一定程度的風(fēng)險(xiǎn)和傷害。尤其是對(duì)嚴(yán)重的、可能危及生命的風(fēng)險(xiǎn),如免疫效應(yīng)細(xì)胞相關(guān)神經(jīng)毒性綜合征、細(xì)胞因子風(fēng)暴和腫瘤溶解綜合征的臨床癥狀和治療措施描述應(yīng)盡可能詳細(xì),對(duì)遠(yuǎn)期風(fēng)險(xiǎn)如兒童受試者生長發(fā)育影響、育齡期受試者生殖和遺傳毒性、基因整合引起的繼發(fā)腫瘤、病毒載體再激活的告知應(yīng)盡可能全面,以便受試者能自主權(quán)衡個(gè)人獲益和潛在社會(huì)獲益,做出個(gè)人最佳判斷,自主自愿參與試驗(yàn)。此外,若項(xiàng)目涉及限制行為能力的受試者,還需征得其法定監(jiān)護(hù)人知情同意,并尊重限制行為能力受試者的治療意向。
3.2.4 細(xì)化風(fēng)險(xiǎn)監(jiān)測與控制措施 CGT 制劑的安全性受細(xì)胞類型、生物學(xué)活性、作用靶點(diǎn)和基因修飾等因素共同影響,不良事件的嚴(yán)重程度和發(fā)生時(shí)間與制劑在體內(nèi)的存續(xù)、增殖與分布等特點(diǎn)密切相關(guān)[13]。CGT 項(xiàng)目開展前,應(yīng)系統(tǒng)收集科學(xué)文獻(xiàn)和研究報(bào)告中同類或相關(guān)產(chǎn)品的安全性信息,對(duì)研究中潛在風(fēng)險(xiǎn)進(jìn)行分析和評(píng)估,制訂可行的風(fēng)險(xiǎn)監(jiān)測計(jì)劃和風(fēng)險(xiǎn)控制預(yù)案,詳細(xì)描述特定風(fēng)險(xiǎn)的識(shí)別、預(yù)防、診斷、治療和預(yù)后隨訪的實(shí)施細(xì)則。CGT 項(xiàng)目應(yīng)制訂完善的不良事件記錄和報(bào)告制度,規(guī)定試驗(yàn)終(中)止規(guī)則和受試者退出標(biāo)準(zhǔn)。另外,還應(yīng)定期組織對(duì)研究者、研究護(hù)士的培訓(xùn)考核,提高醫(yī)護(hù)及時(shí)發(fā)現(xiàn)和妥善處理受試者試驗(yàn)相關(guān)不良事件的能力。IND-CGT 的申辦方和開展 IIT-CGT 的醫(yī)療機(jī)構(gòu)應(yīng)為項(xiàng)目購買臨床試驗(yàn)保險(xiǎn),且保險(xiǎn)的理賠范圍、賠付額度和有效期限能夠覆蓋遲發(fā)性不良反應(yīng)和預(yù)期不良事件的醫(yī)療費(fèi)用。
4 結(jié)語
CGT 制劑在重大疾病領(lǐng)域和罕見病領(lǐng)域已顯示巨大應(yīng)用潛力,有望解決某些疾病療效甚微或無藥可醫(yī)的局面。我國 CGT 領(lǐng)域正在加速崛起,有許多新技術(shù)不斷涌入,也面臨著諸多倫理審查挑戰(zhàn)。在致力于探索 CGT 穩(wěn)定性、有效性、精準(zhǔn)性和安全性的同時(shí),應(yīng)正確認(rèn)識(shí)其中存在的倫理問題,從倫理學(xué)原則出發(fā),進(jìn)一步完善研究基礎(chǔ),選擇合適的研究設(shè)計(jì)方法,規(guī)范知情同意程序,細(xì)化風(fēng)險(xiǎn)監(jiān)測與控制措施,充分保護(hù)受試者生命健康權(quán)益,引導(dǎo)國內(nèi) CGT 臨床研究行業(yè)健康發(fā)展。
猜你喜歡
體育科技文獻(xiàn)通報(bào)(2022年3期)2022-05-23 13:46:54
天津外國語大學(xué)學(xué)報(bào)(2021年3期)2021-08-13 08:32:18
遼金歷史與考古(2021年0期)2021-07-29 01:06:54
科技傳播(2019年22期)2020-01-14 03:06:54
遼金歷史與考古(2019年0期)2020-01-06 07:45:20
民用飛機(jī)設(shè)計(jì)與研究(2019年4期)2019-05-21 07:21:24
電子制作(2018年11期)2018-08-04 03:26:04
汽車工程學(xué)報(bào)(2017年2期)2017-07-05 08:13:02
國際商務(wù)財(cái)會(huì)(2017年8期)2017-06-21 06:14:14
電子制作(2017年23期)2017-02-02 07:17:19
