高效液相色譜法同時測定牙膏中六種氯酚類防腐劑
2024-03-18 04:38:22夏澤敏聶明霞黃敏涵李鑫宇譚建華席紹峰
口腔護理用品工業 2024年1期
關鍵詞:方法
廖 娜 夏澤敏 汪 毅 聶明霞 黃敏涵 李鑫宇 譚建華 席紹峰
(廣州質量監督檢測研究院,廣州 511447)
氯酚類化合物(chlorophenols, CPs)是氯取代苯酚類化合物的總稱,因具有良好的殺菌殺蟲效果,被廣泛用作殺菌劑、防腐劑[1],也被作為殺菌防腐劑廣泛使用在口腔清潔護理產品中。然而,CPs是廣泛的內分泌干擾物,對生物體具有致癌、致畸、致突變的“三致”效應。其性質穩定,難以生物降解,能在環境中相對持久地存在,且易通過食物鏈在生物體內富集[2],會對人體健康造成不利影響,因此多種CPs已被多國列入優先控制的毒性污染物名單。在我國,《牙膏用原料規范(GB 22115-2008)》中對幾種氯酚類防腐劑有明確規定:雙氯酚為牙膏中限用組分,牙膏中最大允許使用濃度為0.5%,并且需在產品標簽中注明“含雙氯酚”;溴氯芬、芐氯酚、氯二甲酚作為牙膏中許用防腐劑,其最大使用濃度分別為0.1%、0.2%、0.5%;六氯酚和p-氯-m-甲酚為牙膏中禁用組分。
通過對國內外相關標準和文獻資料進行檢索,近年來在個人護理產品中CPs的檢測方面,檢測方法主要有高效液相色譜法[3~6]、氣相色譜法[7]和氣相色譜-質譜聯用法[8,],涉及的產品類型主要是化妝品、香皂等皮膚清潔用品。然而,化妝品和牙膏在配方方面存在較大差異,化妝品的方法標準僅可以作為參考,而在分析實際樣品時,樣品的溶解、均質、提取、凈化、測定等操作均不能完全通用。因此,本方法擬采用高效液相色譜法測定牙膏中p-氯-m-甲酚、氯二甲酚、雙氯酚、芐氯酚、 溴氯芬、六氯酚等六種氯酚類防腐劑,為廣大日化行業的檢測機構和企業用戶提供一種快捷簡便、準確高效的測定方法。
1 實驗部分
1.1 儀器與試劑
高效液相色譜儀(美國安捷倫公司);Milli-Q超純水器(美國Millipore公司);IKA MS3 digital渦旋振蕩器(德國IKA公司);SK8200H超聲波清洗器(上海科導超聲儀器有限公司);BS 224S電子天平(德國賽多利斯公司)。
p-氯-m-甲酚(CAS號35421-08-0)、六氯酚(CAS號70-30-4 )、雙氯酚(CAS號97-23-4)、氯二甲酚(CAS號88-04-0)、芐氯酚(CAS號120-32-1)及溴氯芬(CAS號15435-29-7),純度均大于98%,德國Dr.Ehrenstorfer公司;甲醇,色譜純,德國Merck公司;乙腈,色譜純,德國Merck公司;甲酸,分析純,上海安譜科學儀器有限公司;石英砂,分析純,廣州化學試劑廠,超純水,電阻率為18.2 MΩ·cm。
乙腈溶液(70%,v/v):準確移取700mL乙腈置于適量水中,再加水稀釋至1000mL,混勻。
甲酸溶液(0.1%,v/v):準確移取1mL甲酸置于適量水中,再加水稀釋至1000mL,混勻。
1.2 標準溶液的配制
分別準確稱取p-氯-m-甲酚、氯二甲酚、雙氯酚、芐氯酚、溴氯芬及六氯酚的標準品10mg (精確至0.1mg)與10mL容量瓶中,用70%乙腈溶液定容至刻度,配制成質量濃度約為1000mg/L的標準儲備液。使用時,稀釋至所需要的質量濃度的標準工作溶液。
1.3 樣品處理
將牙膏試樣擠出約20mm后,準確稱取試樣1g(精確到0.001g)于50mL聚丙烯離心管中,加入約1g石英砂,渦旋后準確加入70%乙腈溶液20mL,渦旋混勻后超聲提取10min,取適量提取液以10000r/min離心5min,上清液經0.22μm濾膜過濾后待測。
1.4 儀器條件
色譜條件:色譜柱:Weltch XB-C18(150mm×4.6mm,5μm);流動相:A:0.1甲酸,B:乙腈;梯度洗脫程序(0~2min,70%~50% A;2~7min,50% A;7~12min,50%~10% A;12~19min,10% A;19~19.1min,10%~70% A;19. 1~25min,70% A);流速:0.8mL/min;柱溫:30℃;進樣體積:20μL;檢測波長:六氯酚、溴氯芬檢測波長為298nm,p-氯-m-甲酚、氯二甲酚、雙氯酚、芐氯酚檢測波長為282nm。
2 結果與討論
2.1 提取方式及溶解的選擇
根據p-氯-m-甲酚、氯二甲酚、雙氯酚、芐氯酚、溴氯芬及六氯酚等六種氯酚的結構以及理化性質,對提取溶劑和提取方法進行考察,篩選合理的提取方法。六種氯酚皆不溶或微溶于水,易溶于多數有機溶劑,考慮到牙膏產品在水中分散效果較好,本方法采用有機溶劑-水體系作為提取溶劑。考察了不同比例的甲醇水溶液和乙腈水溶液(0%、20%、40%、50%、60%、80%、100%)兩種溶劑體系對含有六種氯酚的提取效果。結果顯示,隨著有機相比例增加,樣液變澄清,樣液變得容易經濾膜過濾,當有機相比例超過90%,樣品容易成團,不易分散。在乙腈水溶劑體系中,不同比例有機溶劑提取的目標化合物回收率相似,皆能達到90%。在甲醇水體系中,隨著甲醇比例增加,回收率逐漸增加,甲醇比例達到70%,回收率高于90%。綜合兩個溶劑體系對六種氯酚的提取情況來看,70%乙腈水溶液的響應面積最高。同時,考慮到牙膏一般具有較好的水分散性,提取溶劑中有機溶劑的比例越高,牙膏在提取溶劑中的分散效果越差。因此選擇70%乙腈水作為六種氯酚的提取溶劑。另外,為了提高牙膏在提取溶劑中的分散效果,本方法選擇加入適量石英砂對牙膏試樣進行輔助分散。
2.2 色譜柱的選擇
考察了Athena C8柱(250mm×4.6mm,5μm)、Eclipse XDB-phenyl(250mm×4.6mm,5μm)以及Xbridge-C18柱(250mm×4.6mm,5μm)、AQ-C18(250 mm×4.6mm,5μm)、Diamonsil -C18(150mm×4.6mm,5μm)、Weltch XB-C18(150mm×4.6mm,5μm)、Proshell EC C18(150mm×4.6mm,5μm)等不同色譜柱對p-氯-m-甲酚、氯二甲酚、雙氯酚、芐氯酚、溴氯芬及六氯酚等六種組分和樣品基質的分離效果。結果顯示,上述色譜柱對標準溶液中目標化合物都可以達到良好的分離效果,但在實際樣品分析時,Weltch XB-C18(150mm×4.6mm,5μm)和Eclipse XDB-phenyl(150mm×4.6mm,5μm)對大部分樣品的分離效果較好,由于溴氯芬及六氯酚在色譜柱上難以洗脫,應避免使用含碳量高的色譜柱。鑒于C18色譜柱通用性好,本方法選定Weltch XB-C18(150mm×4.6mm,5μm)為分析色譜柱。
2.3 流動相的選擇
六種氯酚分子結構中的羥基和色譜柱固定相中殘留的硅羥基存在較強的氫鍵作用,使化合物在色譜柱上保留較強,因此,若僅以甲醇-水或乙腈-水作為流動相,可能會出現色譜峰嚴重拖尾或不出峰的情況。然而,加入甲酸等改性劑,可以有效減小拖尾現象。因此,本方法考察了甲醇-0.1%甲酸水和乙腈-0.1%甲酸水兩種流動相體系。實驗結果表明,在0.1%甲酸-乙腈流動相體系中,六種氯酚的分離度好,峰形對稱,靈敏度高。因此,本方法選用0.1%甲酸-乙腈作為流動相。此外,六種目標物極性差異較大,不適合采用等度洗脫,而使用梯度洗脫時,隨時間增加逐漸增加有機相比例,不僅可以加快目標物的洗脫,還可以減小半峰寬。優化后的六種氯酚標準物質色譜圖見圖1和圖2。
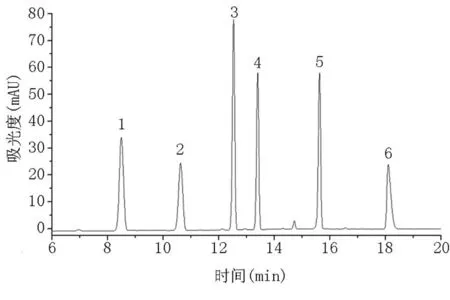
圖1 標準物質溶液的液相色譜圖(λ=282nm)
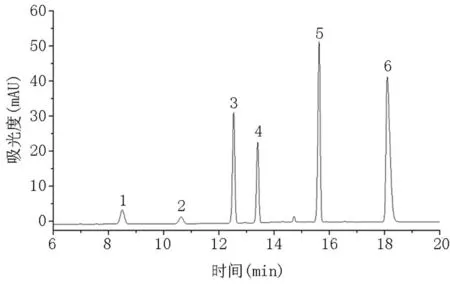
圖2 標準物質溶液的液相色譜圖(λ=298nm)
2.4 檢測波長的選擇
在200~400nm 范圍內對六種氯酚的標準溶液進行全波長掃描,分別獲得相應的紫外吸收光譜圖(圖3所示)。
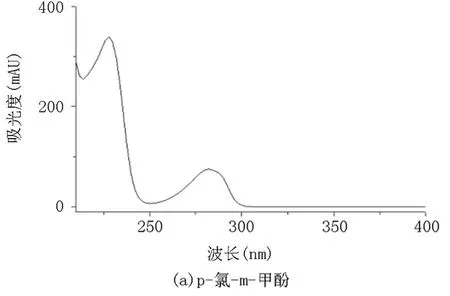
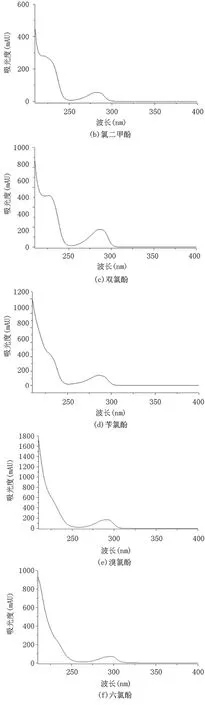
圖3 六種標準物質溶液的紫外吸收光譜圖
由表1可知,p-氯-m-甲酚、氯二甲酚、雙氯酚、芐氯酚、溴氯芬和六氯酚的質量濃度在0.5~20μg/mL范圍內與相應色譜峰面積呈良好的線性關系,六種氯酚的方法檢出限為4μg/g,方法定量限為10μg/g。
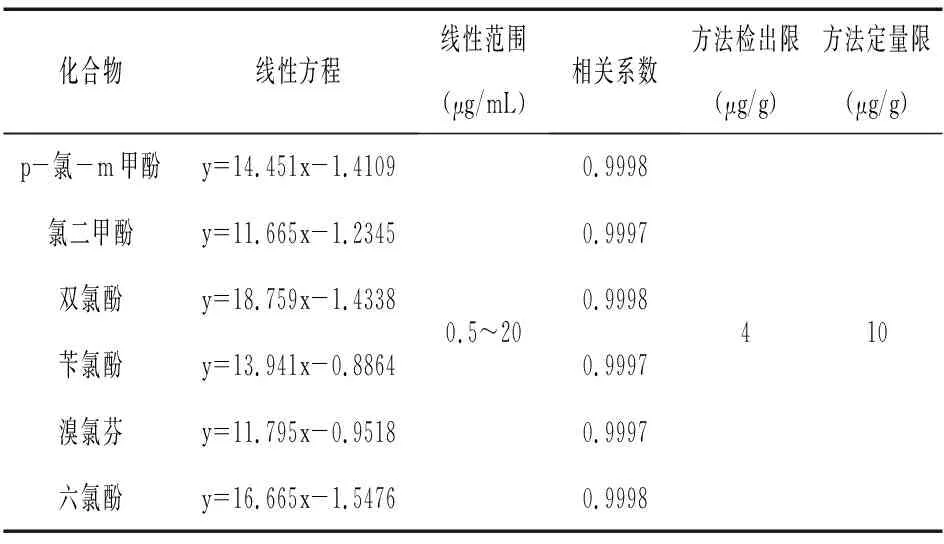
表1 六種目標物的線性方程、線性范圍、相關系數、方法檢出限和定量限
2.5 線性方程和檢出限
在1.4儀器條件下對系列標準工作溶液進行測定。以各目標物質量濃度(x)為橫坐標,對應峰面積(y)為縱坐標,分別繪制標準工作曲線。對陰性樣品添加適量六種氯酚混合標準溶液,按照試樣前處理方法和儀器條件進行測定,以信噪比S/N≥3確定方法檢出限,以信噪比S/N≥10確定方法定量限。線性范圍、線性方程、相關系數、檢出限和定量限見表2。
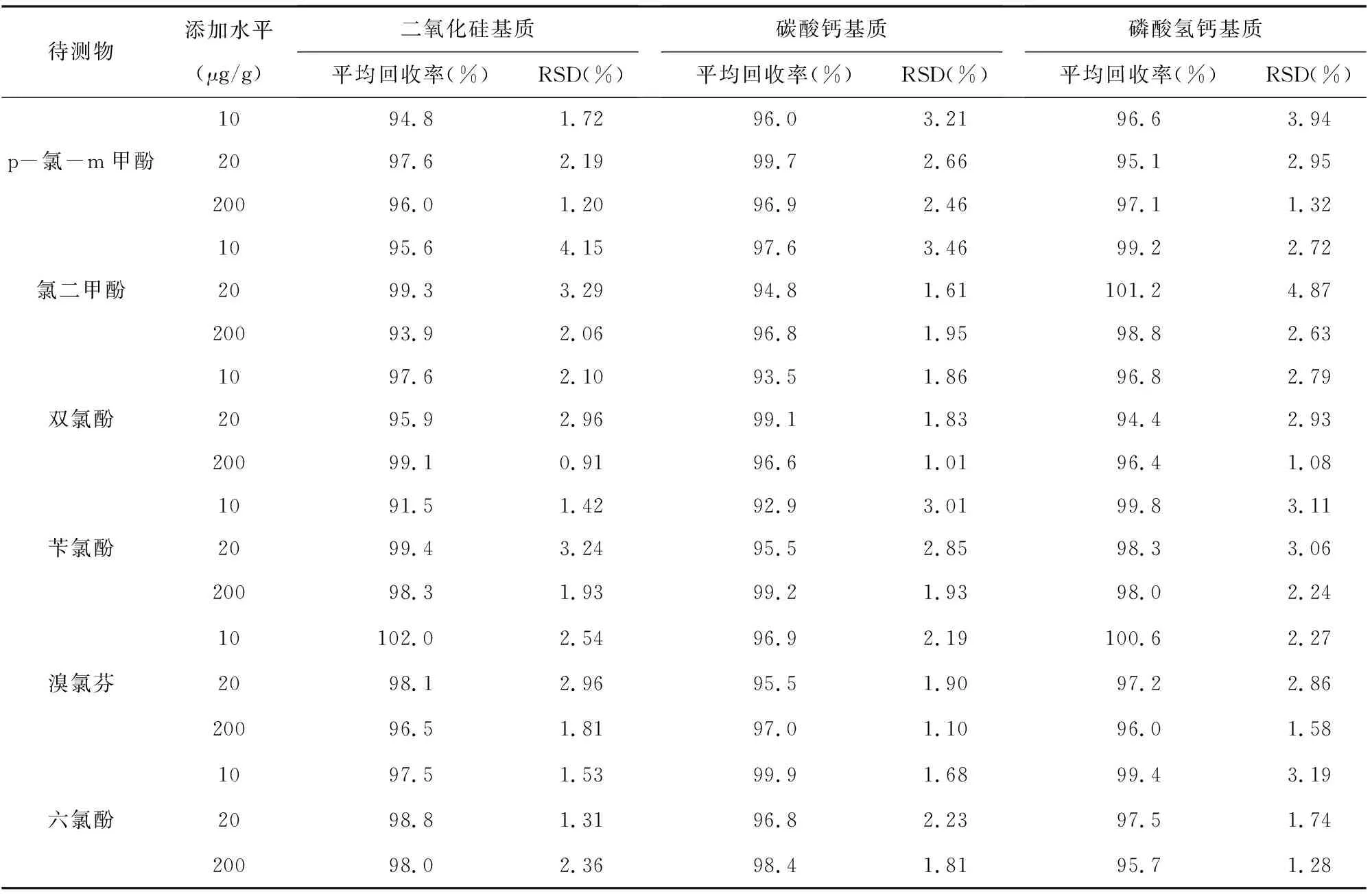
表2 精密度試驗和加標回收試驗結果(n=6)
2.6 精密度試驗及加標回收試驗
選用二氧化硅基質、碳酸鈣基質和磷酸氫鈣基質3種空白牙膏樣品為加標基質,按1.3方法進行處理,分別加入低、中、高3個加標水平的氯酚標準溶液(添加水平分別為10μg/g、20mg/kg、200μg/g),在1.4儀器條件下平行測定6次,測定結果見表2。由表2可知,p-氯-m甲酚的平均回收率為94.8%~99.7%,相對標準偏差為1.20%~3.94%(n=6);氯二甲酚的平均回收率為93.9%~101.2%,相對標準偏差為1.61%~4.87%(n=6);雙氯酚的平均回收率為93.5%~99.1%,相對標準偏差為0.91%~2.96%(n=6);芐氯酚的平均回收率為91.5%~99.8%,相對標準偏差為1.42%~3.24%(n=6);溴氯芬的平均回收率為95.5%~102.0%,相對標準偏差為1.10%~2.96%(n=6);六氯酚的平均回收率為95.7%~99.9%,相對標準偏差為1.28%~3.19%(n=6)。結果表明,本方法具有良好的準確度和精密度,滿足分析要求。
3 結論
本研究采用高效液相色譜技術建立了牙膏中p-氯-m-甲酚、氯二甲酚、雙氯酚、芐氯酚、溴氯芬及六氯酚的檢測方法。對提取條件和色譜條件等進行了優化,并將該方法應用于不同基質牙膏的檢測。結果表明,該方法能對p-氯-m-甲酚、氯二甲酚、雙氯酚、芐氯酚、溴氯芬及六氯酚進行準確測定,且方法精密度好,滿足相關檢測需求,可為牙膏產品的質量監控提供有力的技術支撐。
猜你喜歡
中老年保健(2021年9期)2021-08-24 03:52:04
河北畫報(2021年2期)2021-05-25 02:07:46
中學生數理化(高中版.高考理化)(2020年2期)2020-04-21 05:33:04
兒童繪本(2020年5期)2020-04-07 17:46:30
兒童故事畫報(2019年5期)2019-05-26 14:26:14
Coco薇(2016年2期)2016-03-22 02:42:52
山東青年(2016年1期)2016-02-28 14:25:23
Coco薇(2015年1期)2015-08-13 02:47:34
小雪花·成長指南(2015年7期)2015-08-11 15:03:12
小雪花·成長指南(2015年4期)2015-05-19 14:47:56
