大豆類病變皺葉突變體NT301遺傳分析和2對基因定位
2024-03-28 02:36:30王亞琪徐海風李曙光傅蒙蒙余希文趙志鑫楊加銀趙團結
作物學報 2024年4期
關鍵詞:大豆
王亞琪 徐海風 李曙光 傅蒙蒙 余希文 趙志鑫 楊加銀,* 趙團結
大豆類病變皺葉突變體遺傳分析和2對基因定位
王亞琪1徐海風1李曙光1傅蒙蒙1余希文1趙志鑫1楊加銀1,*趙團結2,*
1江蘇徐淮地區淮陰農業科學研究所 / 淮安市農業生物技術重點實驗室 / 農業農村部淮河下游種質創制重點實驗室, 江蘇淮安 223001;2南京農業大學大豆研究所/ 國家大豆改良中心(南京) / 農業農村部大豆生物學與遺傳育種重點實驗室(綜合) / 作物遺傳與種質創新國家重點實驗室, 江蘇南京 210095
通過研究類病變突變體, 挖掘抗病基因, 利用分子設計育種的方法快速培育優良抗病大豆新品種, 可減輕化學農藥對環境的污染, 降低病害的抗藥性。本研究以60Coγ誘變獲得的類病變皺葉突變體為父本, 分別與W82、KF1和KF35進行雜交, 構建了F2和F2:3分離群體, 通過SSR標記和SNP分析, 將目標基因1 ()縮小到18號染色體937 kb區間內, 包含66個候選基因, 將目標基因2 ()縮小到8號染色體130 kb區間內, 包含15個候選基因。接著, 本研究利用基因芯片技術對近等基因系進行了基因表達譜研究, 得到了差異表達基因參與的KEGG調控通路。另外對8號染色體的15個候選基因進行半定量與熒光定量RT-PCR表達分析發現, 只有基因在突變體與野生型中的差異達到了4倍, 推測基因是突變體的候選基因。
大豆; 類病變突變體; 皺葉; 基因表達譜
大豆含有豐富優質的蛋白和油脂, 是重要的糧食作物和經濟作物, 在我國糧食和飼料結構中占有重要位置。而我國大豆生產正面臨嚴峻的形勢, 隨著對大豆需求逐年增加, 供求矛盾日益突出, 如今已經成為世界最大的大豆進口國[1-2]。病害嚴重影響大豆產量和品質, 培育優良抗病品種, 既節約了資源, 又可減少化學農藥對環境的污染, 還能降低病害的抗藥性。而分子設計育種是解決這一困境的重要途徑, 加強基礎研究, 挖掘重要性狀的關鍵基因及其調控網絡, 是分子設計育種的關鍵。
近年來的許多研究表明, 類病變突變體病斑的形成與過敏反應及程序性細胞死亡有關, 是研究植物抗病分子機制的良好材料。類病變突變體是指植物組織上自發形成細胞死亡, 與外部環境的脅迫沒有直接關系, 往往形成病斑或過敏反應[3-4]。1997年, 研究人員首先在玉米和擬南芥中鑒定到了控制類病變性狀的基因[5-6], 后來在番茄和水稻中也鑒定到了類病變的候選基因[7-8]。Wang等[9]通過圖位克隆的方法從大豆類病變突變體中鑒定出候選基因脂氫過氧化物裂解酶HPL, 能夠響應病蟲害脅迫。Ma等[10]鑒定出一個大豆光依賴的類病變突變體, 其候選基因編碼一種共型卟啉原III氧化酶GmLMM2, 參與四吡咯生物合成, 突變體增強了對大豆疫霉的抗性。這些研究揭示了大多數類病變突變體能夠激活防御反應、甚至增強了對病原菌的抗性, 這些基因涉及多個途徑: 如抗病信號途徑、植物激素及防衛信號分子、程序性細胞死亡失控、葉綠素合成和脂類代謝等。
類病變皺葉突變體主要特點為葉脈與葉肉細胞發育不協調, 產生與病毒感染相似的形態特征, 葉面積大大減小, 結實率降低, 而大豆中的研究并不深入。Stephens等[11]從再生植株中發現了一個不穩定遺傳的皺葉突變體, 后代分離不穩定, 屬于細胞質遺傳, 往往出現嵌合體。Wilcox和Abney[12]從M2中鑒定到一個隱性遺傳的葉寬變窄的皺葉突變體, 遺傳解析表明由單基因控制。聶智星等[13]從60Coγ誘變的大豆突變體庫中篩選到一個皺葉突變體, 葉緣卷曲, 類似花葉病毒感染。在普通菜豆(L.)中, 異源雜交后代出現了雙隱性基因控制的皺葉突變體[14]。Song等[15]鑒定到一個大豆皺葉突變體, 小葉頂端壞死, 而小葉其他部分繼續生長, 葉面積和葉片干重顯著降低。Ochar等[16]報道了一個EMS誘變的大豆皺葉突變體, 編碼具有CCT結構域的B-box型鋅指蛋白突變導致葉片皺縮, 株高降低。
基因重復或多倍化在植物進化過程中普遍存在,且不同的進化階段都有發生。全基因組復制是重復基因最重要的來源之一, 其中一部分基因消亡, 一部分基因被保留但功能發生了變化, 這些對進化起了重要作用[17]。大豆基因組在過去的6000萬年時間里至少經歷了2輪全基因組復制, 存在大量的重復序列, 研究者推測這些重復基因的功能也發生了變化[18], 研究重復基因的功能對研究進化起到了重要作用。之前的研究中, 我們發現了一個新的類病變突變體, 葉片皺縮, 株高降低, 對F2代和F2:3家系的遺傳分析表明該性狀被2對隱性基因控制, 功能冗余, 并且將這2對基因定位在18號和8號染色體上。
本研究將目標基因1 ()縮小到18號染色體937 kb區間, 目標基因2 ()縮小到8號染色體130 kb區間, 包含15個候選基因。為進一步縮小候選基因的范圍, 本研究對8號染色體的15個候選基因進行半定量與實時熒光定量表達分析發現, 只有基因在突變體與野生型中的差異達到了4倍, 推測基因是突變體的候選基因。
1 材料與方法
1.1 遺傳材料獲得與構建
大豆品種玉譽(Tamahomore)、Williams 82 (W82)、科豐1號(Kefeng 1, KF1)、科豐35 (Kefeng 35, KF35)等種植資源均來自于南京農業大學國家大豆改良中心。突變體來自于60Coγ誘變的玉譽[19],并經過多代回交及自交, 得到近等基因系的野生型(wild type, WT)和突變型(mutant type, MT)。然后分別以W82、KF1和KF35為母本, 以為父本構建雜交群體, W82×雜交組合F2代分離群體共1912株, F2:3代株系共156行; KF1×雜交組合F2代分離群體共451株, F2:3代株系共182行; KF35×雜交組合F2代分離群體共362株, F2:3代株系共176行。
1.2 石蠟切片觀察葉脈結構
在V4至V5時期, 采集野生型和突變體植株頂部比較幼嫩的葉片, 采用常規石蠟切片法制片。用手術刀片切成大小0.5~1.0 cm左右的組織, FAA固定液固定, 保證組織表面無氣泡, 1 d后轉移至70%乙醇中, 按照不同濃度的乙醇脫水, 然后用二甲苯透明劑透明, 用石蠟浸蠟和包埋, 切片機切片, 粘附劑粘片與烤片, 再用二甲苯脫蠟, 不同濃度梯度的酒精復水, 用番紅-固綠對染, 再經過不同濃度梯度的酒精脫水、二甲苯透明和中性樹膠封固, 在LEICA DMLB型顯微鏡下觀察并照相。
1.3 目的基因定位
對于質量性狀, 根據孟德爾遺傳定律來進行遺傳分析。由于突變體第1片三出復葉就表現皺葉, 肉眼很容易分辨, 因此, 用肉眼觀測進行表型調查。調查F1、F2和F2:3代(F2單株衍生的株行)的表型, 統計其性狀分離比, 用卡平方適合性測驗計算其顯著性。以隱性遺傳為例, 如果F1代單株全部表現為正常, F2代單株出現正常︰皺葉=3︰1, 表現正常F2單株衍生的F2:3代株行正常不分離行︰分離行=1︰2, 則為隱性單基因遺傳。如果F1代單株全部表現為正常, F2代單株出現正常︰皺葉=15︰1, 表現正常F2單株衍生的F2:3代株行正常不分離行︰分離行=7︰8, 則為隱性雙基因遺傳。
采用SSR標記和SNP連鎖定位的方法進行基因定位。對F2和F2:3群體, V3至V4期突變體皺葉表型出現時, 調查表型并取頂部幼嫩葉片, 用CTAB法提取DNA。根據Song等[20]開發的大豆全基因組微衛星標記(SSR)對初定位區間進行標記加密, 當定位區間還有交換單株時, 就要開發新的SSR標記, 進一步縮小定位區間。方法如下: 在SoyBase (https:// soybase.org/)中下載W82參考基因組目標區段序列, 利用SSRHunter[21]篩選SSR標記, 參數設置優先選擇3~6個堿基的重復序列, 并根據軟件所給的結果, 利用NCBI Primer-BLAST (https://www.ncbi.nlm.nih. gov/tools/primer-blast/index.cgi?LINK_LOC=BlastHome)在上下游150 bp內設計引物, 保證擴增產物長度在100~300 bp之間, 上下游引物m值相差不大于5℃, 由通用生物(安徽)股份有限公司合成。PCR擴增利用2×Master Mix (P112-01, 諾唯贊生物科技股份有限公司, 南京)進行, 用8%的聚丙烯酰胺凝膠(PAGE)對PCR產物進行電泳檢測和銀染顯色[19]。
1.4 基因表達譜試驗流程及數據分析
當突變體出現皺葉表型時, 混樣取野生型和突變體的葉片, 迅速裝入凍存管, 并置于液氮中, 以備提取大豆總RNA, 野生型和突變體各3個生物學重復, 送上海伯豪生物公司進行基因表達譜分析。流程是: 首先, 將檢測合格的RNA樣本進行純化, 再反轉錄合成第1鏈、第2鏈cDNA, 然后利用雙鏈cDNA合成生物素標記的aRNA, 純化后與芯片雜交,進行測序分析。
1.5 候選基因預測及半定量、定量RT-PCR檢測
在SoyBase網站下載定位區間內的所有基因ID, 根據Phytozome (http://phytozome-next.jgi.doe.gov/)和SoyKB (https://soykb.org/)網站的基因功能注釋, 預測候選基因, 并下載基因組及轉錄組的序列, 利用NCBI Primer-BLAST設計半定量和實時熒光定量PCR引物(附表1), 檢測定位區間內基因的表達水平。V3至V4時期, 取頂部完全展開的嫩葉(每個材料3個生物學重復), 迅速放入液氮冷凍, 再用總RNA提取試劑盒(DP419, 天根生化科技有限公司, 北京)提取總RNA, 檢測其濃度及質量, 并稀釋至終濃度為5 ngmL–1, 用反轉錄試劑盒(R223, 諾唯贊生物科技股份有限公司, 南京)將RNA反轉為cDNA。半定量RT-PCR以cDNA為模板, 在普通PCR儀進行, 根據瓊脂糖凝膠電泳檢測結果確定了內參基因和目的基因以26個循環為最佳。實時熒光定量PCR反應以cDNA為模板, 用染料法試劑盒(Q311, 諾唯贊生物科技股份有限公司, 南京), 在Roche Light Cycler 480 II上進行, 相對表達量采用2–ΔΔCt計算方法。半定量和熒光定量RT-PCR每個樣本3個技術重復。
2 結果與分析
2.1 突變體NT301的形態特點與遺傳分析
之前的研究已經詳細報道了突變體的形態特征[19], 該突變體植株矮小, 結實率低, 葉片表現出感染花葉病毒癥狀(圖1-B, D), 種子大小與野生型無明顯差異, 石蠟切片顯示皺葉葉片橫切面的內部結構(圖1-F)。突變體表皮細胞排列紊亂, 不規則凸起, 致使表面形成褶皺, 表皮細胞和維管束之間的厚角組織層數增多, 導致葉脈寬度變窄, 木質部細胞變小, 葉肉細胞單位面積的細胞數目異常增多, 柵欄組織和海綿組織連在一起, 中間幾乎沒有空隙。田間調查雜交組合后代表型并進行遺傳分析。由表1可知, KF1×群體F1代葉片形態表現正常, F2代451個單株中, 有430株表現正常, 21株表現皺葉, 卡方測驗符合15︰1的分離比(=0.48), F2:3代182個株系中, 有88個株系表現正常不分離, 94個株系出現分離, 卡方測驗符合7︰8的分離比(=0.70)。KF35×群體F1代葉片形態表現正常, F2代362個單株中, 有342株表現正常, 20株表現皺葉, 卡方測驗符合15︰1的分離比(=0.80), F2:3代176個株系中, 有87個株系表現正常不分離, 89個株系出現分離, 卡方測驗符合7︰8的分離比(=0.51)。已報道過W82×群體F2代單株正常︰皺葉符合15︰1的分離比(=0.57), F2:3代株系正常不分離︰分離符合7︰8的分離比(=0.84)[19]。表明皺葉性狀受2對隱性基因控制。

圖1 突變體NT301的形態特征
A: 苗期野生型植株; B: 苗期植株; C: 開花期野生型植株; D: 開花期植株; E: 野生型葉片橫切面; F:葉片橫切面。標尺: 1 cm (A, B); 5 cm (C, D); 200 μm (E, F)。p: 柵欄組織; s: 海綿組織; vb: 維管束。
A: wild-type plants at seedling stage; B: mutantat seedling stage; C: wild-type plants at flowering stage; D: mutantatflowering stage; E: the transverse section of wild-type leaves; F: the transverse section ofleaves. Bar: 1 cm (A, B); 5 cm (C, D); 200 μm (E, F). p: palisade parenchyma; s: spongy parenchyma; vb: vascular bundle.
2.2 突變體NT301皺葉基因定位
在之前的報道中, 將的目標基因定位在了18號染色體BARCSOYSSR_18_0415和BARCSOYSSR_18_0485兩個SSR標記之間, 物理距離1.85 Mb, 將目標基因定位在了8號染色體BARCSOYSSR_08_1700和Satt409之間, 物理距離1.18 Mb。為了進一步精細定位, 利用KF1×群體F2:3家系277個隱性單株在18號染色體定位區間內加密了3個標記, 分別是18_0419、18_0444和18_0481, 用這3個標記分別檢測出了10個、5個和3個交換單株; 利用W82×群體F2:3家系327個隱性單株加密了2個標記, 分別是18_0419和18_0430, 并檢測了標記18_0485, 用這3個標記分別檢測出了9個、3個和17個交換單株, 綜合2個群體的定位結果, 將定位在了18_0444和18_0481之間, 物理距離937 kb。利用KF1×群體F2:3家系262個隱性單株在8號染色體定位區間內加密了3個標記, 分別是08_1724、08_1736和08_1753, 并檢測了標記08_1800, 用這4個標記分別檢測出了52個、33個、9個和23個交換單株; 利用W82×群體F2:3家系315個隱性單株加密了5個SSR標記和3個SNP變異位點, 分別是08_1724、08_1738、08_1748、SSR18、SSR40和SNP1、SNP2、SNP3, 用這些標記分別檢測出了10個、10個、10個、2個、2個和2個、1個、2個交換單株, 綜合2個群體的定位結果, 將定位在了SNP2和SNP3之間, 物理距離130 kb (圖2)。根據SoyBase和Phytozome網站的基因注釋信息, 18號和8號染色體目標區段內分別有66個和15個候選基因。
2.3 突變體NT301皺葉候選基因參與的調控網絡
利用大豆Affymetrix基因組芯片檢測皺葉突變體與野生型葉片組織的表達差異, 該芯片包含37,500個大豆()的轉錄本, 背景值小于100, 檢出率為40%以上表明芯片、樣本及操作均達到合格標準。在本研究中, 野生型樣品1-1、1-2、1-3和突變體樣品2-1、2-2、2-3的探針檢出率均大于40% (表2), 表明試驗數據比較可靠。
采用隨機方差模型對差異基因進行篩選, 共獲得了3568個差異表達基因, 其中上調2337個, 下調1231個。采用選擇Cluster 3.0軟件對差異基因進行聚類, 將功能顯著差異的600個基因使用hierarchical, average linkage算法進行分析(圖3), 野生型和突變體能夠明顯分為2類, 說明芯片數據的重復性較好。
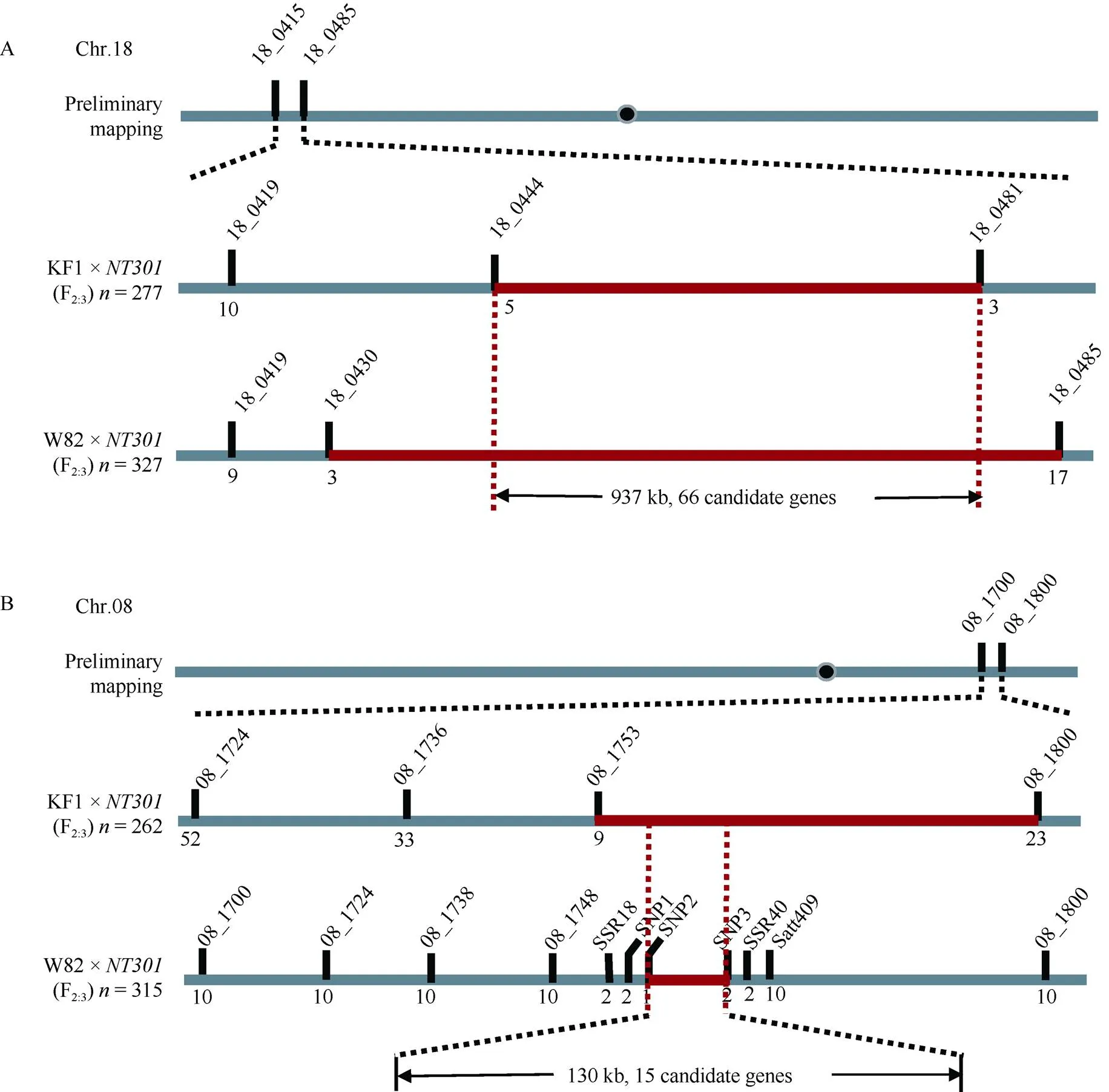
圖2 突變體NT301 2對候選基因定位
A: 18號染色體定位; B: 8號染色體精細定位。
A: mapping ofon chromosome 18; B: fine mapping ofon chromosome 8.
整合SoyBase、Phytozome等數據庫中的大豆基因組和蛋白組信息, 結合eggNOG-mapper (http:// eggnog-mapper.embl.de/)對差異表達基因進行Pathway注釋, 利用KEGG (https://www.genome. jp/kegg/)數據庫進行富集分析, 篩選出差異基因顯著影響的Pathway (圖4)。上調基因所顯著參與的合成代謝通路共15個(<0.05, 圖4-A), 包括9個代謝通路、5個合成通路和1個植物與病原菌互作通路, 前5條通路達到了極顯著水平(0.01)。谷胱甘肽代謝通路基因功能集中在谷胱甘肽轉移酶, 如谷胱甘肽S轉移酶18/20/22、類γ-谷氨酰轉肽酶1; 苯丙素生物合成和苯丙氨酸代謝通路基因功能集中在類過氧化物酶, 如類過氧化物酶4/12/52/73, 有一些涉及病原菌誘導; 纈氨酸、亮氨酸和異亮氨酸降解通路基因功能集中在酰基輔酶A; 氮代謝通路基因功能集中在谷氨酰胺合成酶。下調基因所顯著參與的合成代謝通路共有10個(<0.05, 圖4-B), 包括8個代謝通路和2個合成通路, 前6條代謝通路達到了極顯著水平。卟啉和葉綠素代謝通路基因功能集中在卟啉原脫氨酶、脫羧酶和氧化酶等; 氨基糖和核苷酸糖代謝通路基因功能集中在葉綠體戊糖、己糖激酶、糖苷酶等; 次生代謝產物的生物合成基因功能集中在葉綠體卟啉脫羧酶、轉移酶, 糖基合酶、轉移酶等, 轉移酶、氨酰-tRNA合成基因功能集中在氨酰-tRNA合成酶, 淀粉和蔗糖代謝基因功能集中在葉綠體半乳糖醛酸海藻糖磷酸合酶、果膠酯酶等; 糖酵解/糖異生代謝通路基因功能集中在己糖激酶。

圖3 突變體NT301與野生型差異表達基因的聚類圖
橫坐標代表樣品名稱及樣品的聚類結果, 縱坐標代表差異基因及基因的聚類結果。S1為突變體, S2為野生型, 各3個重復, 不同的行代表不同的基因。顏色代表了基因在樣品中的表達量水平。
The horizontal axis represents the sample names and clustering results of the samples, while the vertical axis represents the differentially expressed genes and clustering results of the genes. S1 represents the mutant, and S2 represents the wild type, three times repeat. Different rows represent different genes. Color represents the relative expression level of genes in the samples.
2.4 突變體NT301皺葉候選基因預測
根據生物信息學預測, 18號和8號染色體目標區段內分別有66個和15個候選基因, Phytozome網站上對8號染色體的候選基因功能注釋如表3所示。為了進一步預測候選基因, 取V3至V4時期大豆葉片, 提取總RNA, 做半定量和熒光定量PCR, 比較分析候選基因在野生型和突變體葉片中的表達水平。結果如圖5所示, 只有基因在突變體與野生型中的差異達到了4倍, 推測基因是突變體的候選基因, 功能注釋為UDP糖基轉移酶超家族蛋白。將18號和8號染色體定位區間內所有基因的氨基酸序列做了聚類分析, 結果顯示8號染色體候選基因與18號染色體聚類在一起, 序列相似度最高, 推測是18號染色體定位區間內的候選基因(附圖1)。
在擬南芥中的同源基因是, 編碼UDP鼠李糖-花青素-3-葡萄糖苷鼠李糖基轉移酶, 是類黃酮合成通路的下游基因, 能夠催化花青素苷生成花色素類, 進而在谷胱甘肽轉移酶的作用下生成花青素谷胱甘肽。研究認為, 類黃酮基因的表達與生長素極性運輸呈負相關, 抑制了生長素轉運蛋白在芽和莖中的表達, 從而抑制了腋芽的生長[22]。突變體候選基因的高表達導致下游谷胱甘肽也隨之升高, 這與表達譜芯片的結果一致, 由此影響了上游類黃酮基因的表達, 使生長素運輸受到影響, 最終導致葉脈與葉肉細胞發育不協調, 形成皺葉的表型。
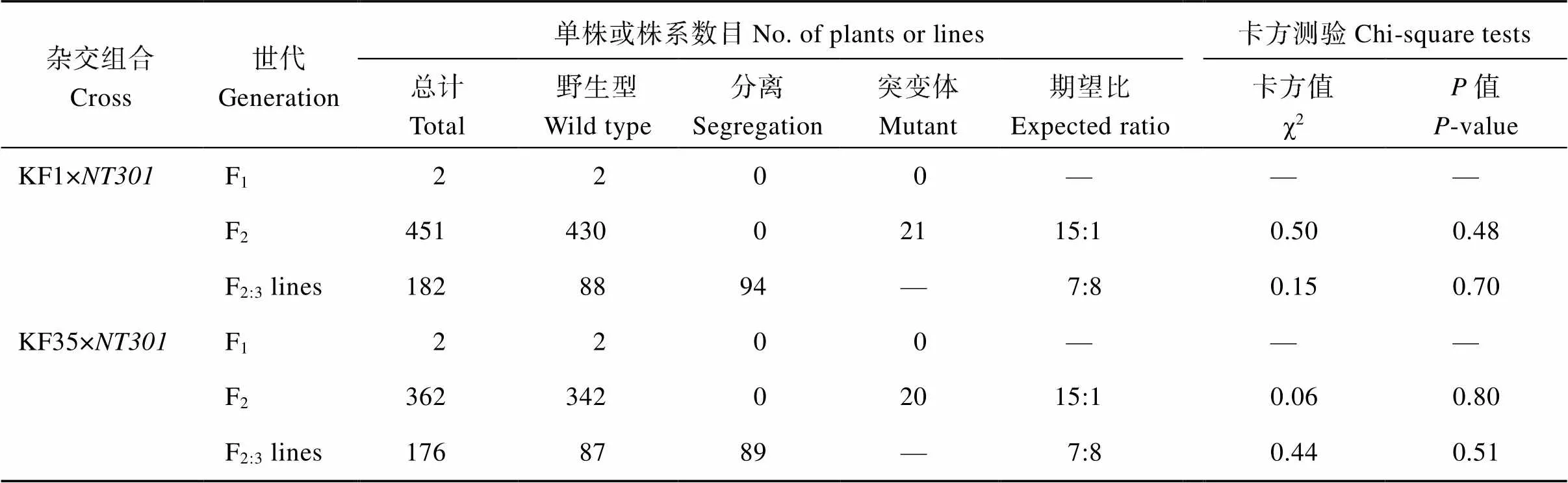
表1 雜交組合F2和F2:3群體野生型和突變體植株分離率適合性檢驗
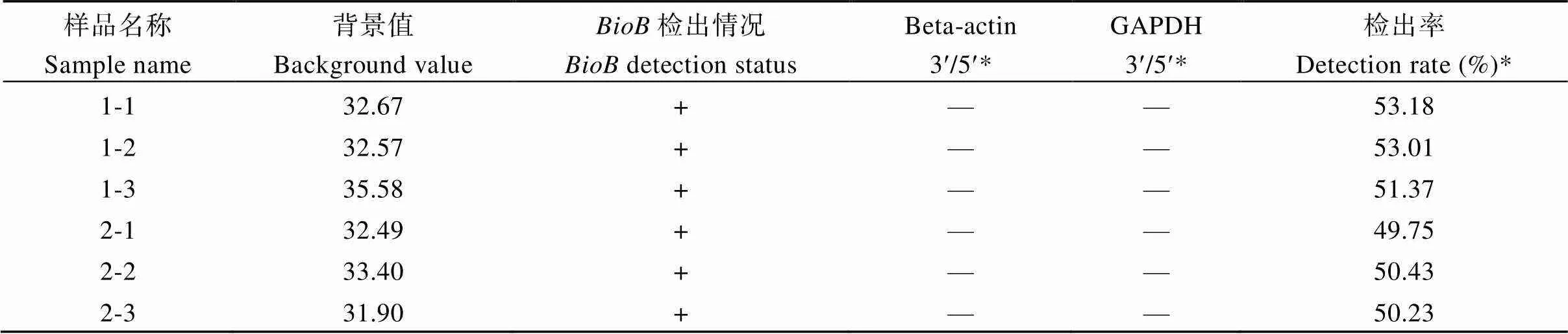
表2 芯片試驗質控情況
Beta-actin 3¢/5¢*與GAPDH 3¢/5¢*: 管家基因3¢端信號與5¢端信號比值至少有一個不大于3, 此標準僅適用于以下8種表達譜芯片: 人、小鼠、大鼠、斑馬魚、線蟲、果蠅、酵母和大腸桿菌表達譜芯片。檢出率Detection rate (%)*: 檢出點總數與全部探針數的比值為該芯片的檢出率。
Beta-actin 3¢/5¢* and GAPDH 3¢/5¢*: the ratio of the 3' ends signal to the 5' ends signal of the housekeeping genes should not exceed 3. This standard only applies to the following 8 expression profile chips: human, mouse, rat, zebrafish, nematode, fruit fly, yeast, andexpression profile chips. Detection rate (%)*: the ratio of the total number of detected probes to the total number of probes is the detection rate of the chip.
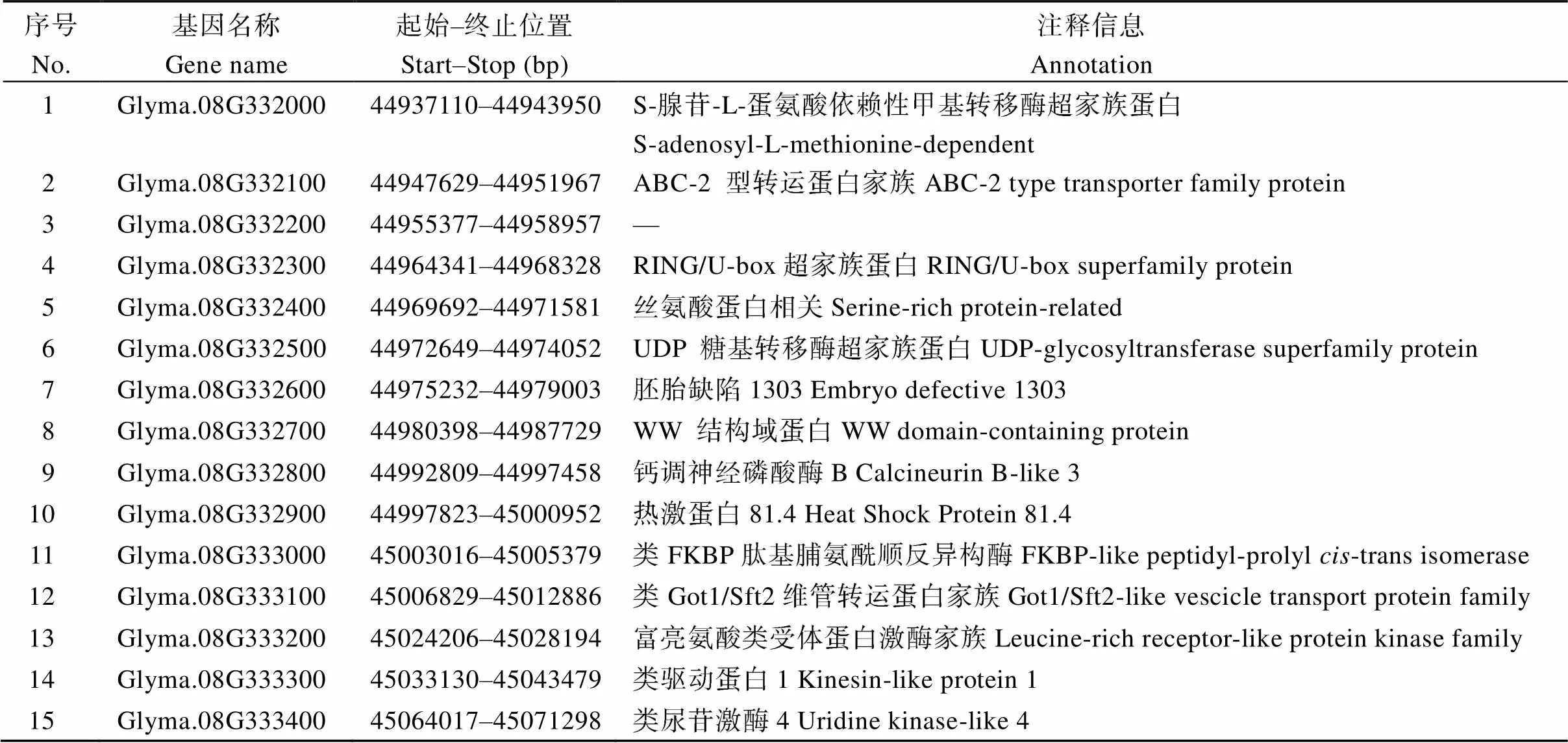
表3 rl2位點候選基因功能注釋
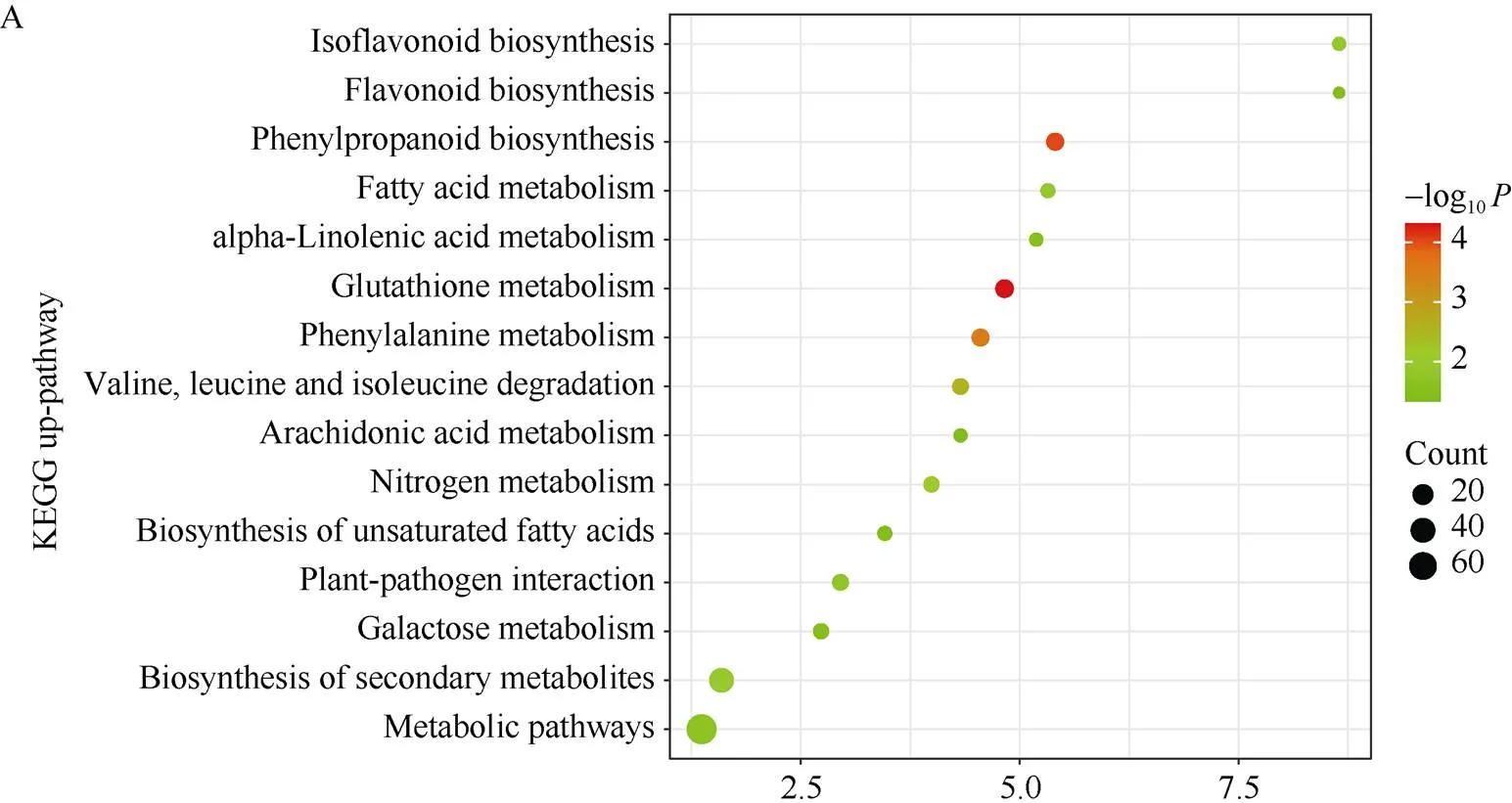
(圖4)
A: 上調基因富集的KEGG通路; B: 下調基因富集的KEGG通路。圖中–log10越大, 表示該通路越顯著。橫坐標表示富集值, 縱坐標表示富集的通路。圓圈大小表示基因數量。
A: the enrichment of upregulated genes in the KEGG; B: the enrichment of downregulated genes in the KEGG. The figure shows that the –log10is greater, the pathway is more significant. The horizontal axis represents the enrichment value, and the vertical axis represents the pathway of enrichment. The size of the circle indicates the number of genes.
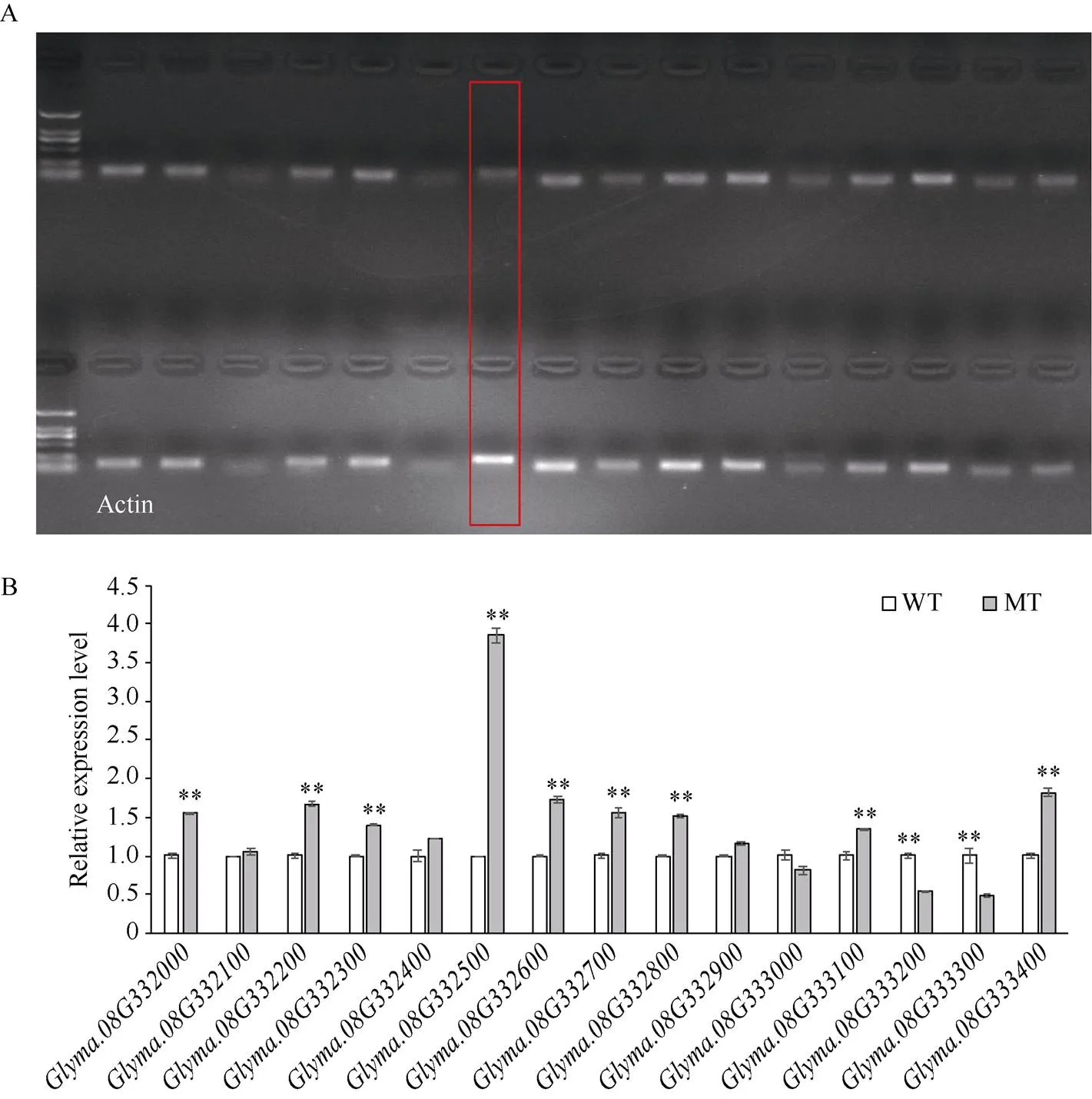
圖5 8號染色體定位區間內基因的表達分析
A: 8號染色體定位區間內基因半定量PCR分析; B: 8號染色體定位區間內基因熒光定量PCR分析。紅色方框表示野生型和突變體表達量差異最大的基因, 采用學生測驗進行顯著性檢驗(**:≤ 0.01), 誤差線表示標準差(= 3)。
A: the semi quantitative PCR analysis of genes in the mapping interval of chromosome 8; B: the quantitative RT-PCR analysis of genes in the mapping interval of chromosome 8. The red box indicates the most differentially expressed gene between wild type and mutant. The significant test was carried out by the student’s-test (**:≤ 0.01). The error bars indicate the SDs (= 3).
3 討論
葉片皺縮受2對隱性基因的控制, 分別位于18號和8號染色體上, 只有2對基因同時突變為隱性時, 才出現類似花葉病毒感染的特征, 其中任何一對基因為顯性時, 都不出現花葉病毒的癥狀, 所以推測這2對基因是同源基因且功能冗余。根據SoyBase網站參考基因組的序列信息, 18號染色體定位區間內有兩段序列的重復序列剛好位于8號染色體的定位區間之內, 且此區域內8號染色體的基因密度大于18號染色體。根據前人的研究結果, 由于不對稱進化, 18號染色體著絲粒及周圍區域與8號染色體短臂有大量的同源基因, 全基因組復制事件之后, 這些基因經歷了從常色狀態到異色狀態的完全或部分切換, 或者剛好相反[23], 因此推測和基因也是由同源基因進化而來的。
在大豆基因組測序沒有完成之前, 研究者利用經典遺傳學中同源基因的遺傳特性及基因定位能夠推斷其同源連鎖群, 這對于研究大豆的進化是十分有利的。Lohnes等[24-25]揭示了和位點共同調控子葉顏色的遺傳機制, 只有2個位點都為隱性即時, 子葉為綠色, 其余基因型都為黃色, 并且驗證了和所在連鎖群為部分同源連鎖群。Fang等[26]進一步研究發現,和與擬南芥和水稻中的SGR (STAY-GREEN)基因是同源基因, 與本研究不同的是,與在衰老早期均抑制了葉綠素的降解, 但在衰老后期的抑制作用沒有強烈, 表明基因與基因的功能并非完全冗余。
為適應進化, 多倍體植物中重復基因可能發生假基因化、亞功能化或新功能化[27], Ji等[28]認為染色體或片段重復在基因家族的擴張過程中起到了重要作用。重復基因往往在轉錄水平上有差異, 從而引起表型的變化[29]。大豆生長習性基因()有4個同源基因, 但功能已經發生了變化, 它們在不同發育階段有不同的表達模式, 而且在群體中的變異位點也是不同的[30]。多倍體由于增加了遺傳變異和復制基因的緩沖作用, 對極端環境具有更強的耐受性, Chen等[31]篩選到一個新的既受到除草劑選擇也發生了多倍化擴增的基因, 并驗證了該基因與氰氟草酯和惡唑酰草胺的結構相互作用, 從分子水平揭示了多倍體雜草具有更強的除草劑適應性的原因。
到目前為止, 大豆中對于類病變皺葉突變體相關基因挖掘較少, 其所在代謝通路上下游關系還不明確。而擬南芥中研究較多, 如過表達或者下調其靶基因, 影響茉莉酸合成通路, 促進細胞增殖或者葉片皺縮[32]。擬南芥膽色素原脫氨酶缺失導致葉片發育畸形, 細胞壞死, 生長受到抑制, 開花延遲, 并觸發植物防御機制激活[33]。擬南芥突變體葉脈扭曲并開叉形成雙脈, 并且側根異常彎曲, 細胞非正常擴張, 游離生長素水平正常, 但是運輸受到損害[34]。擬南芥和突變體葉脈不能形成網狀結構, 變成沿主脈的狹長一條, 且形狀不規則, 葉緣有缺刻, 作者推測是由于生長素分布不均勻導致的[35]。葉脈與葉肉發育不協調, 葉緣形成不規則褶皺, 葉面積大大減小, 而葉肉細胞密度大大增加, 推測可能跟生長素分配不均有關。
4 結論
本研究利用大豆類病變皺葉突變體與W82、KF1、KF35構建的F2、F2:3雜交群體, 用SSR和SNP等分子標記進行基因定位, 將目的基因1 ()縮小到18號染色體937 kb區間內, 包含66個候選基因, 將目的基因2 ()縮小到8號染色體130 kb區間內, 包含15個候選基因, 并根據表達譜芯片和qPCR的結果, 推測是候選基因。
[1] 呂慧穎, 王道文, 葛毅強, 魏珣, 鄧向東, 楊維才, 田志喜. 大豆育種行業創新動態. 植物遺傳資源學報, 2018, 19: 464–467. Lyu H Y, Wang D W, Ge Y Q, Wei X, Deng X D, Yang W C, Tian Z X. Innovation of soybean breeding industry.,2018, 19: 464–467 (in Chinese with English abstract).
[2] 田志喜, 劉寶輝, 楊艷萍, 李明, 姚遠, 任小波, 薛勇彪. 我國大豆分子設計育種成果與展望. 中國科學院院刊, 2018, 33: 915–922. Tian Z X, Liu B H, Yang Y P, Li M, Yao Y, Ren X B, Xue Y B. Update and prospect of soybean molecular module-based designer breeding in China.,2018, 33: 915–922 (in Chinese with English abstract).
[3] Huang Q N, Yang Y, Shi Y F, Chen J, Wu J L. Spotted-leaf mutants of rice ()., 2010, 17: 247–256.
[4] Wu C J, Bordeos A, Madamba M R S, Baraoidan M, Ramos M, Wang G L, Leach J E, Leung H. Rice lesion mimic mutants with enhanced resistance to diseases., 2008, 279: 605–619.
[5] Gray J, Close P S, Briggs S P, Johal G S. A novel suppressor of cell death in plants encoded by thegene of maize., 1997, 89: 25–31.
[6] Dietrich R A, Richberg M H, Schmidt R, Gean C, Dangl J L. A novel zinc finger protein is encoded by thegene and functions as a negative regulator of plant cell death., 1997, 88: 685–694.
[7] Tang X, Xie M, Kim Y J, Zhou J, Klessig D F, Martin G B. Overexpression ofactivates defense responses and confers broad resistance., 1999, 11: 15–29.
[8] Yamanouchi U, Yano M, Lin H, Yamada K. A rice spotted leaf gene,, encodes a heat stress transcription factor protein., 2002, 99: 7530–7535.
[9] Wang Y, Liu M, Ge D, Bhat J A, Li Y, Kong J, Liu K, Zhao T. Hydroperoxide lyase modulates defense response and confers lesion-mimic leaf phenotype in soybean ((L.) Merr.)., 2020, 104: 1315–1333.
[10] Ma J, Yang S, Wang D, Tang K, Feng X. Genetic mapping of a light-dependent lesion mimic mutant reveals the function of coproporphyrinogen iii oxidase homolog in soybean., 2020, 11: e557.
[11] Stephens P A, Barwale U B, Nickell C D, Widholm J M. A cytoplasmically inherited, wrinkled-leaf mutant in soybean., 1991, 82: 71–73.
[12] Wilcox J R, Abney T S. Inheritance of a narrow, rugose-leaf mutant in., 1991, 82: 421–423.
[13] 聶智星, 代金英, 吉家正, 陳薇, 趙團結. 大豆葉突變體的發掘與特性分析. 江蘇農業科學, 2013, 41(1): 86–88.Nie Z X, Dai J Y, Ji J Z, Chen W, Zhao T J. Identification and characterization of soybean leaf mutant.,2013, 41(1): 86–88 (in Chinese).
[14] Singh S P, Molina A. Inheritance of crippled trifoliolate leaves occurring in interracial crosses of common bean and its relationship with hybrid dwarfism., 1996, 87: 464–469.
[15] Song X, Wei H, Cheng W, Yang S, Zhao Y, Li X, Luo D, Zhang H, Feng X. Development of INDEL markers for genetic mapping based on whole genome resequencing in soybean., 2015, 5: 2793–2799.
[16] Ochar K, Bo-Hong S U, Zhou M M, Liu Z X, Gao H W, Flamlom S, Qiu L J. Identification of the genetic locus associated with the crinkled leaf phenotype in a soybean (L.) mutant by BSA-Seq technology., 2022, 21: 3524–3539.
[17] 孫紅正, 葛頌. 重復基因的進化: 回顧與進展. 植物學報, 2010, 45: 13–22. Sun H Z, Ge S. Review the evolution of duplicate genes., 2010, 45: 13–22 (in Chinese with English abstract).
[18] Schmutz J, Cannon S B, Schlueter J, Ma J, Jackson S A. Genome sequence of the palaeopolyploid soybean., 2010, 463: 178–183.
[19] Wang Y, Chen W, Zhang Y, Liu M, Kong J, Yu Z, Jaffer A M, Gai J, Zhao T. Identification of two duplicated loci controlling a disease-like rugose leaf phenotype in soybean., 2016, 56: 1611–1618.
[20] Song Q J, Jia G F, Zhu Y L, Grant D, Nelson R T, Hwang E Y, Hyten D L, Cregan P B. Abundance of SSR motifs and development of candidate polymorphic SSR markers (BARCSOYSSR_ 1.0) in soybean., 2010, 50: 1950–1960.
[21] 李強, 萬建民. SSRHunter, 一個本地化的SSR位點搜索軟件的開發. 遺傳, 2005, 27: 808–810. Li Q, Wan J M. SSRHunter, development of a local searching software for SSR sites., 2005, 27: 808–810 (in Chinese with English abstract).
[22] Lazar G, Goodman H M., a regulator of the flavonoid pathway, controls vegetative axillary bud outgrowth in., 2006, 103: 472–476.
[23] Du J, Tian Z, Sui Y, Zhao M, Song Q, Cannon S B, Cregan P, Ma J. Pericentromeric effects shape the patterns of divergence, retention, and expression of duplicated genes in the paleopolyploid soybean., 2012, 24: 21–32.
[24] Lohnes D G, Specht J E, Cregan P B. Evidence for homoeologous linkage groups in the soybean., 1997, 37: 254–257.
[25] Chao W S, Liu V, Thomson W W, Platt K, Walling L L. The impact of chlorophyll-retention mutations,and, during embryogeny in soybean., 1995, 107: 253–262.
[26] Fang C, Li C, Li W, Wang Z, Zhou Z, Shen Y, Tian Z. Concerted evolution ofandto regulate chlorophyll degradation in soybean., 2014, 77: 700–712.
[27] Innan H, Kondrashov F. The evolution of gene duplications: classifying and distinguishing between models., 2010, 11: 97–108.
[28] Ji Q, Zhang L S, Wang Y F, Wang J. Genome-wide analysis of basic leucine zipper transcription factor families in,and., 2009, 13: 174–182.
[29] Wang Y P, Wang X Y, Paterson A H. Genome and gene duplications and gene expression divergence: a view from plants., 2012, 1256: 1–14.
[30] Tian Z X, Wang X, Lee R, Li Y, Specht J E, Nelson R L, McClean P E, Qiu L, Ma J. Artificial selection for determinate growth habitin soybean., 2010, 107: 8563–8568.
[31] Chen K, Yang H, Peng Y, Liu D, Zhang J, Zhao Z, Wu L, Lin T, Bai L. Genomic analyses provide insights into the polyploidization-driven herbicide adaptation inweeds., 2023, 21: 1642–1658.
[32] Schommer C, Palatnik J F, Aggarwal P, Chetelat A, Cubas A, Farmer E E, Nath U, Weigel D. Control of jasmonate biosynthesis and senescence bytargets., 2008, 6: e230.
[33] Quesada V, Sarmiento-Ma?ús R, González-Bayón R, Hricová A, Ponce M R, Micol J L. PORPHOBILINOGEN DEAMINASE deficiency alters vegetative and reproductive development and causes lesions in., 2013, 8: e53378.
[34] Carland F M, McHale N A.: a gene involved in auxin transport and vascular patterning in., 1996, 122: 1811–1819.
[35] Cnops G, Neyt P, Raes J, Petrarulo M, Nelissen H, Malenica N, Luschnig C, Tietz O, Ditengou F, Palme K, Azmi A, Prinsen E, Lijsebettensa M V. Theandgenes function in several patterning processes during early leaf deve-lopment in., 2006, 18: 852–866.
附表1 本研究所用到的引物
Table S1 Primers used in this study
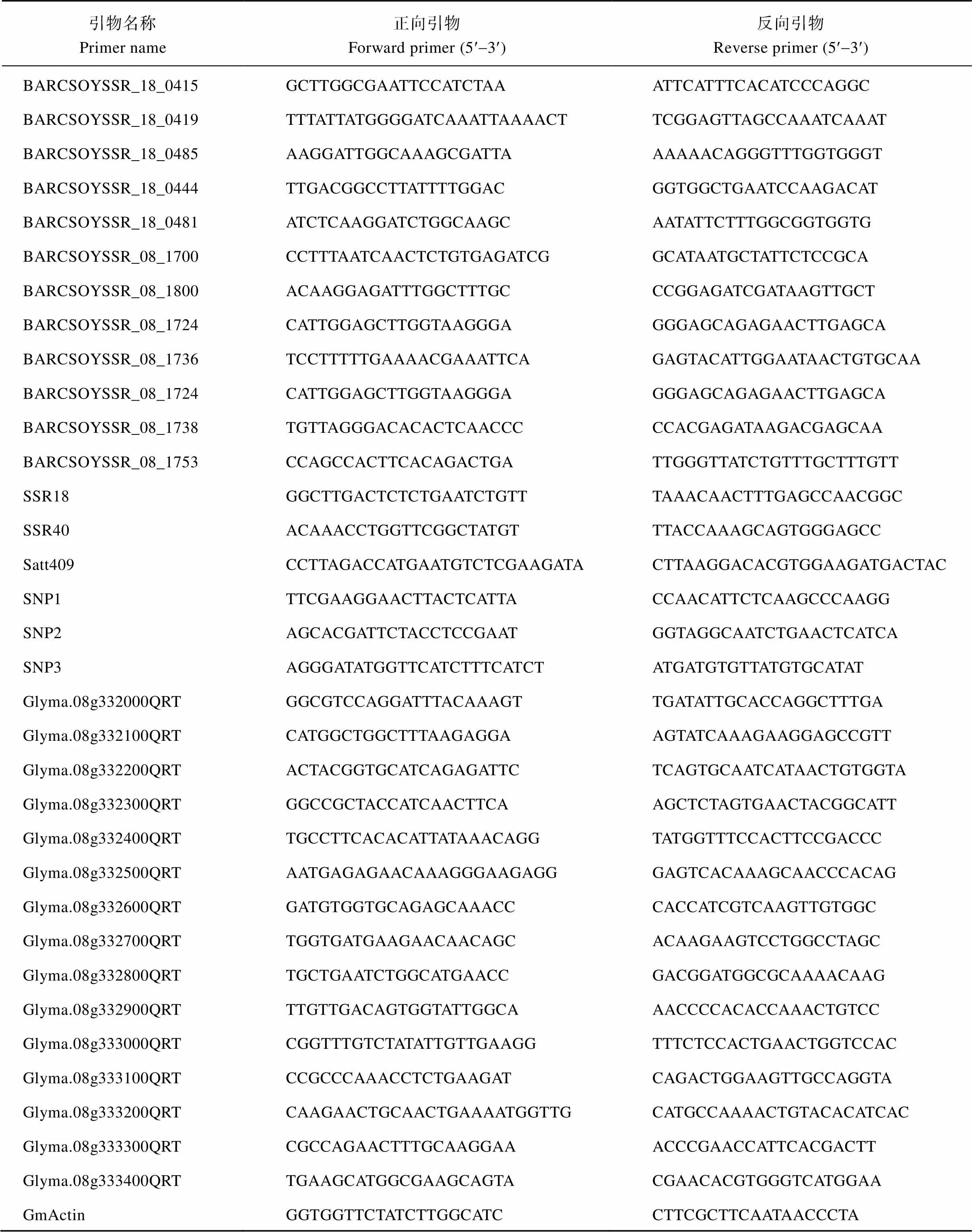
引物名稱Primer name正向引物Forward primer (5¢-3¢)反向引物Reverse primer (5¢-3¢) BARCSOYSSR_18_0415GCTTGGCGAATTCCATCTAAATTCATTTCACATCCCAGGC BARCSOYSSR_18_0419TTTATTATGGGGATCAAATTAAAACTTCGGAGTTAGCCAAATCAAAT BARCSOYSSR_18_0485AAGGATTGGCAAAGCGATTAAAAAACAGGGTTTGGTGGGT BARCSOYSSR_18_0444TTGACGGCCTTATTTTGGACGGTGGCTGAATCCAAGACAT BARCSOYSSR_18_0481ATCTCAAGGATCTGGCAAGCAATATTCTTTGGCGGTGGTG BARCSOYSSR_08_1700CCTTTAATCAACTCTGTGAGATCGGCATAATGCTATTCTCCGCA BARCSOYSSR_08_1800ACAAGGAGATTTGGCTTTGCCCGGAGATCGATAAGTTGCT BARCSOYSSR_08_1724CATTGGAGCTTGGTAAGGGAGGGAGCAGAGAACTTGAGCA BARCSOYSSR_08_1736TCCTTTTTGAAAACGAAATTCAGAGTACATTGGAATAACTGTGCAA BARCSOYSSR_08_1724CATTGGAGCTTGGTAAGGGAGGGAGCAGAGAACTTGAGCA BARCSOYSSR_08_1738TGTTAGGGACACACTCAACCCCCACGAGATAAGACGAGCAA BARCSOYSSR_08_1753CCAGCCACTTCACAGACTGATTGGGTTATCTGTTTGCTTTGTT SSR18GGCTTGACTCTCTGAATCTGTTTAAACAACTTTGAGCCAACGGC SSR40ACAAACCTGGTTCGGCTATGTTTACCAAAGCAGTGGGAGCC Satt409CCTTAGACCATGAATGTCTCGAAGATACTTAAGGACACGTGGAAGATGACTAC SNP1TTCGAAGGAACTTACTCATTACCAACATTCTCAAGCCCAAGG SNP2AGCACGATTCTACCTCCGAATGGTAGGCAATCTGAACTCATCA SNP3AGGGATATGGTTCATCTTTCATCTATGATGTGTTATGTGCATAT Glyma.08g332000QRTGGCGTCCAGGATTTACAAAGTTGATATTGCACCAGGCTTTGA Glyma.08g332100QRTCATGGCTGGCTTTAAGAGGAAGTATCAAAGAAGGAGCCGTT Glyma.08g332200QRTACTACGGTGCATCAGAGATTCTCAGTGCAATCATAACTGTGGTA Glyma.08g332300QRTGGCCGCTACCATCAACTTCAAGCTCTAGTGAACTACGGCATT Glyma.08g332400QRTTGCCTTCACACATTATAAACAGGTATGGTTTCCACTTCCGACCC Glyma.08g332500QRTAATGAGAGAACAAAGGGAAGAGGGAGTCACAAAGCAACCCACAG Glyma.08g332600QRTGATGTGGTGCAGAGCAAACCCACCATCGTCAAGTTGTGGC Glyma.08g332700QRTTGGTGATGAAGAACAACAGCACAAGAAGTCCTGGCCTAGC Glyma.08g332800QRTTGCTGAATCTGGCATGAACCGACGGATGGCGCAAAACAAG Glyma.08g332900QRTTTGTTGACAGTGGTATTGGCAAACCCCACACCAAACTGTCC Glyma.08g333000QRTCGGTTTGTCTATATTGTTGAAGGTTTCTCCACTGAACTGGTCCAC Glyma.08g333100QRTCCGCCCAAACCTCTGAAGATCAGACTGGAAGTTGCCAGGTA Glyma.08g333200QRTCAAGAACTGCAACTGAAAATGGTTGCATGCCAAAACTGTACACATCAC Glyma.08g333300QRTCGCCAGAACTTTGCAAGGAAACCCGAACCATTCACGACTT Glyma.08g333400QRTTGAAGCATGGCGAAGCAGTACGAACACGTGGGTCATGGAA GmActinGGTGGTTCTATCTTGGCATCCTTCGCTTCAATAACCCTA
附圖1 突變體18號和8號染色體定位區間內候選基因氨基酸序列聚類分析
Fig. S1 Clustering analysis of amino acid sequences of candidate genes within the mapping intervals on chromosomes 18 and 8 in the mutant
紅色方框表示8號染色體表達量差異最大的基因和18號染色體的同源基因。
The red box indicates the most differentially expressed geneon chromosome 8 and its homologous geneon chromosome 18.
Genetic analysis and two pairs of genes mapping in soybean mutantwith disease-like rugose leaf
WANG Ya-Qi1, XU Hai-Feng1, LI Shu-Guang1, FU Meng-Meng1, YU Xi-Wen1, ZHAO Zhi-Xin1, YANG Jia-Yin1,*, and ZHAO Tuan-Jie2,*
1Huaiyin Institute of Agricultural Sciences of Xuhuai Region in Jiangsu / Huai’an Key Laboratory for Agricultural Biotechnology / Key Laboratory of Germplasm Innovation in Lower Reaches of the Huaihe River, Ministry of Agriculture and Rural Affairs, Huai’an 223001, Jiangsu, China;2Soybean Research Institute, Nanjing Agricultural University / National Center for Soybean Improvement (Nanjing) / Key Laboratory for Biology and Genetic Improvement of Soybean (General), Ministry of Agriculture and Rural Affairs / National Key Laboratory of Crop Genetics and Germplasm Enhancement, Nanjing 210095, Jiangsu, China
Research on lesion mimic mutant, mining resistance genes, and developing superior disease-resistant new soybean varieties by molecular design breeding methods can contribute to the alleviating the environmental pollution caused by chemical pesticides and drug resistance to disease. In this study, the disease-like rugose leaf mutantobtained by60Coγ mutagenesis as the male parent was crossed with W82, KF1, and KF35, respectively, to construct F2and F2:3segregating populations. Using SSR and SNP markers, target gene 1 () was narrowed to 937 kb on chromosome 18 with 66 genes and target gene 2 () was narrowed to 130 kb on chromosome 8 with 15 genes. The gene expression patterns of the wild type andwere compared using gene chip technology, and the KEGG pathways of the differentially expressed genes were assessed. Moreover, semi quantitative and quantitative RT-PCR methods were used to analyze the relative expression levels of candidate genes on chromosome 8. The results showed that the relative expression level ofinwas four times higher than the wild type. In contrast, the expression levels of other genes showed no more than double difference. Therefore, we suggest thatmay be a candidate gene for.
soybean; disease-like mutant; rugose leaf; gene expression profiles
10.3724/SP.J.1006.2024.34106
本研究由國家自然科學基金項目(32201729), 淮安市自然科學研究計劃項目(聯合專項, HABL202120), 淮安市農業科學研究院高層次引進人才科研啟動發展基金(0112023014B), 淮安市農業科學研究院科研發展基金(HNY202221), 江蘇省種業振興揭榜掛帥項目(JBGS[2021]057)和江蘇省現代作物生產協同創新中心項目(JCIC-MCP)資助。
The study was supported by the National Natural Science Foundation of China (32201729), the Natural Science Research Program of Huai’an (Joint Special Project, HABL202120), the Scientific Research Fund of Startup and Development for Introduced High-level Talents, Huai’an Academy of Agricultural Sciences (0112023014B), the Research and Development Fund Project of Huai’an Academy of Agricultural Sciences (HNY202221), the Core Technology Development for Breeding Program of Jiangsu Province (JBGS[2021]057), and the Jiangsu Collaborative Innovation Center for Modern Crop Production (JCIC-MCP) Program.
趙團結, E-mail: tjzhao@njau.edu.cn; 楊加銀, E-mail: hynksyjy@163.com
E-mail: yqwang_01@126.com
2023-06-27;
2023-10-23;
2023-11-14.
URL: https://link.cnki.net/urlid/11.1809.S.20231113.1429.005
This is an open access article under the CC BY-NC-ND license (http://creativecommons.org/licenses/by-nc-nd/4.0/).
猜你喜歡
農業科技通訊(2023年1期)2023-02-12 07:09:18
今日農業(2022年16期)2022-11-09 23:18:44
中國化肥信息(2022年7期)2022-08-31 01:29:28
中國化肥信息(2022年5期)2022-08-30 01:58:26
今日農業(2021年20期)2021-11-26 01:23:56
今日農業(2021年14期)2021-10-14 08:35:34
下一代英才(酷炫少年)(2018年6期)2018-07-09 03:17:44
農產品市場周刊(2017年4期)2017-03-03 19:40:05
兒童故事畫報·智力大王(2015年10期)2016-01-27 01:01:35
讀寫算(中)(2015年10期)2015-11-07 07:24:12
