超高效液相串聯三重四極桿質譜法測定果凍中胭脂紅酸
2024-04-29 00:00:00汪強
食品安全導刊 2024年1期
關鍵詞:方法

摘 要:建立了超高效液相色譜串聯三重四極桿質譜儀測定果凍中胭脂紅酸含量的方法。樣品通過鹽酸水溶液酸化提取,并經過HLB固相萃取柱凈化后,上機測定,基質外標法定量。胭脂紅酸在5~200 ng·mL-1線性關系良好,相關系數>0.999,方法的檢出限為3.36 ng·g-1,定量限為11.21 ng·g-1。在不同濃度的加標水平下,回收率為91.2%~98.2%,相對標準偏差均<2%。本方法污染小,操作簡便,結果準確,適用于果凍中胭脂紅酸的檢測。
關鍵詞:胭脂紅酸;果凍;超高效液相色譜串聯三重四極桿質譜法
Determination of Carminic Acid in Jelly by Ultra High Performance Liquid Chromatography-Triple Quadrupole Tandem Mass Spectrometry
WANG Qiang
(Maanshan Institute for Food and Drug Control and Adverse Drug Reaction, Maanshan 243000, China)
Abstract: A method for the determination of carminic acid content in jelly using ultra high performance liquid chromatography-triple quadrupole tandem mass spectrometer was established. The sample was acidified and extracted with hydrochloric acid aqueous solution, and after purification with an HLB solid-phase extraction column, it was measured on an instrument and quantified by the matrix external standard method. The linearity of carminic acid was good in the range of 5~200 ng·mL-1 with the correlation coefficient>0.999. The limit of detection of the method was 3.36 ng·g-1, and the limit of quantification was 11.21 ng·g-1. The recoveries ranged from 91.2% to 98.2% at the spiked levels of different concentrations, and the relative standard deviations were all<2%. The method is characterized by low contamination, simple operation, accurate results and suitable for the determination of carminic acid in jelly.
Keywords: carminic acid; jelly; ultra performance liquid chromatography-triple quadrupole tandem mass spectrometry
胭脂紅酸為胭脂蟲紅的主要成分[1],具有蒽醌類結構[2]。胭脂蟲紅具有無毒、耐熱、抗光和染色能力極強等特性,其應用涵蓋食品、化妝品、醫藥等領域[3-5]。胭脂蟲紅雖為天然色素,但也會引發過敏和兒童多動癥等不良反應[6],因此對胭脂蟲紅的使用需要加強監督。目前檢測胭脂紅酸的方法有薄層色譜法[7]、毛細管電泳法[8]、高效液相色譜法[9]、超高效液相色譜串聯三重四極桿質譜法(Ultra High Performance Liquid Chromatography-Triple Quadrupole Tandem Mass Spectrometry,UPLC-MS/MS)[10]等。薄層色譜法、毛細管電泳法和高效液相色譜法對于復雜樣品難以分離目標物,檢測結果不準確[11]。目前UPLC-MS/MS法檢測胭脂紅酸仍有不足之處,如操作復雜、試劑耗材多、凈化效果不理想等,對此本文優化了前處理方法和色譜條件,可以簡單準確地檢測胭脂紅酸的含量,為市場監管提供的技術支持,保障食品安全。
1 材料與方法
1.1 儀器與設備
Arium-Pro型純水機,Sartori-us(德國);ME204E/02型天平(萬分之一),METTLER TOLEDO(瑞士);T25型分散機,IKA(德國);SepLine-S4型全自動固相萃取裝置,萊伯泰科(美國);DMT-2500型多管渦旋混合儀,米歐儀器(中國);SHA-BA型水浴恒溫振蕩器,諾基儀器(中國);N-EVAP5085型氮吹儀,Organomation(美國);HC-3018型臺式高速離心機,中科中佳(中國);0.22 μm微孔濾膜,安譜(中國);3500型超高效液相色譜儀串聯三重四極桿質譜聯用儀,AB(美國)。
1.2 材料與試劑
胭脂紅酸標準物質,由DR提供;甲醇(質譜純),賽默飛;鹽酸(分析純),阿拉丁;甲酸(質譜純),賽默飛;實驗室用水均為超純水。
HLB固相萃取柱:200 mg、6 mL(60 μm)(納譜科技),分別使用6 mL甲醇和6 mL水進行活化。
1.3 實驗方法
1.3.1 溶液配制
鹽酸水溶液(2 mol·L-1):量取84 mL鹽酸至400 mL水中,混勻后,移入500 mL容量瓶中并用水定容至刻度線,搖勻。
胭脂紅酸儲備液(1.0 mg·mL-1):稱取0.100 0 g標準物質,用甲醇溶解,并用甲醇定容至100 mL,配制成質量濃度為1.0 mg·mL-1的標準儲備溶液,4 ℃下避光保存。
胭脂紅酸中間液(1.0 μg·mL-1):移取100 μL"1.0 mg·mL-1胭脂紅酸儲備液,至100 mL容量瓶中,并用2%甲酸水溶液-甲醇(1+1,v/v)稀釋并定容至刻度線。
胭脂紅酸標準工作液:分別移取1.0 μg·mL-1胭脂紅酸中間液5 μL、10 μL、50 μL、100 μL和200 μL至2 g(精確至0.000 1 g)的空白樣品中,配成帶基質的5 ng·mL-1、10 ng·mL-1、50 ng·mL-1、100 ng·mL-1和200 ng·mL-1的胭脂紅酸標準工作液。
1.3.2 樣品前處理
(1)提取。稱取2 g樣品(精確至0.000 1 g)于50 mL離心管中,加入20 mL 2 mol·L-1鹽酸水溶液,使用分散機10 000 r·min-1勻漿3 min后,于100 ℃振搖30 min。之后冷卻至室溫,超聲提取10 min,10 000 r·min-1離心10 min,上清液移入100 mL容量瓶中。重復提取一次,合并上清液,用水定容至刻度,搖勻,備用。
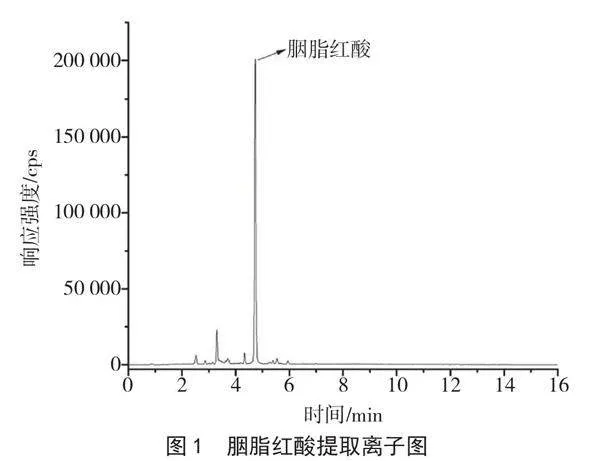
(2)凈化。準確移取10 mL上述備用液,至活化后的HLB固相萃取柱中,控制流速為1 mL·min-1,用12 mL水進行淋洗,抽干,用9 mL甲醇洗脫。洗脫液于40 ℃水浴中氮吹至近干,使用1.0 mL 2%甲酸水溶液-甲醇(1+1,v/v)進行復溶,過0.22 μm微孔濾膜,供UPLC-MS/MS上機測定,所得圖譜見圖1。
1.3.3 色譜條件
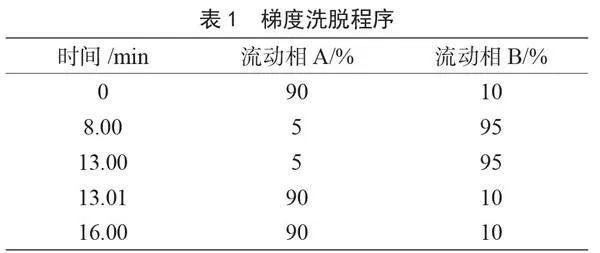
色譜柱:C18柱(150 mm×3.0 mm,3 μm);柱溫:35 ℃;流速:0.5 mL·min-1;進樣體積:10 μL;流動相A:0.3%甲酸水溶液;流動相B:甲醇;梯度洗脫程序見表1。
1.3.4 質譜條件
掃描模式:負離子模式;監測模式:MRM;電噴霧電壓:-4 500 V;離子源溫度:650 ℃;霧化氣壓:55 psi;輔助氣壓:55 psi;氣簾氣壓力:40 psi;碰撞氣壓力:9 psi。胭脂紅酸的質譜參數見表2。
2 結果與分析
2.1 加熱溫度的選擇
本文選擇25 ℃、50 ℃、100 ℃ 3個溫度作為加熱提取的溫度,按照1.3實驗方法進行測定。
25 ℃、50 ℃、100℃鹽酸水溶液的回收率分別為15%、50%、96%。依據回收率效果,選擇100 ℃作為加熱提取的溫度。
2.2 固相萃取柱的選擇
實驗過程中比較聚酰胺SPE柱、混合型強陰離子SPE柱、HLB SPE柱3種固相萃取柱的凈化效果,按照1.3實驗方法進行測定。聚酰胺SPE柱、混合型強陰離子SPE柱、HLB SPE柱的回收率分別為32%、57%、96%。由于聚酰胺SPE柱和混合型強陰離子SPE柱的洗脫液分別用到氨水[11]和磷酸,在氮氣濃縮的過程中,隨著有機試劑的揮發,氨水和磷酸濃度增加,使得目標物分解從而導致回收率低下。依據回收率結果,選用HLB SPE柱作為凈化柱。
2.3 流動相的優化
對比了甲酸水-酸化乙腈、乙酸銨-甲醇、甲酸水-甲醇作為流動相對待測物的影響,結果表明3種流動相對胭脂紅酸色譜峰形、響應值、分離度的影響較小,從試劑的毒性和配制方便性考慮,最終選擇甲酸水-甲醇作為本次實驗的流動相。
2.4 方法的線性范圍、檢出限、定量限
胭脂紅酸在5~200 ng·mL-1濃度范圍內與峰面積具有良好的線性關系,回歸方程為y=3 548.918 35x+5 674.592 75,相關系數>0.999。以3倍S/N(信噪比)和10倍S/N(信噪比)對應的含量作為方法的檢出限和定量限,得到本方法的檢出限為3.36 ng·g-1,定量限為11.21 ng·g-1。
2.5 方法的準確度和精密度
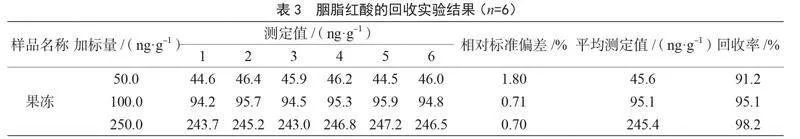
如表3所示,對空白樣品進行3個水平的加標,樣品在50.0 ng·g-1、100.0 ng·g-1、250.0 ng·g-1加標水平下,回收率為91.2%~98.2%,相對標準偏差為0.70%~1.80%(n=6),表明方法的精密度和準確度良好,滿足方法要求。
3 結論
本文采用鹽酸水溶液酸化提取色譜樣品,HLB SPE小柱凈化、濃縮,建立了超高效液相色譜串聯三重四極桿質譜聯用法測定果凍中胭脂紅酸的分析方法。本方法有機試劑用量少、操作簡便、結果準確,不僅為市場監管提供了技術支持,保障人們的食品安全,也為食品生產企業胭脂紅酸質量控制提供了參考。
參考文獻
[1]國家衛生和計劃生育委員會.食品安全國家標準 食品添加劑使用標準:GB 2760—2014[S].北京:中國標準出版社,2014.
[2]林欽.超高效液相色譜法快速檢測食品中胭脂蟲紅酸的含量[J].分析試驗室,2011,30(8):33-35.
[3]鄭小嚴.進口天然色素胭脂蟲紅安全性檢測與分析[J].食品研究與開發,2010,31(4):177-181.
[4]張建云,李志國,趙杰軍,等.中國胭脂蟲發展現狀概述[J].西南農業學報,2007,20(3):560-564.
[5]陳毅怡,劉曉靜,曾曉房,等.食用天然紅色素的研究進展[J].廣州化工,2017,45(23):6-8.
[6]湯沈楊,陳夢瑤,肖花美,等.胭脂蟲及胭脂蟲紅色素的應用研究進展[J].應用昆蟲學報,2019,56(5):969-981.
[7]李明元.反相TLC/光密度掃描分析法食品中蟲膠色素和胭脂蟲紅色素的分析[J].口岸衛生控制,2001(2):45.
[8]許元紅,唐亞軍,吳明嘉.毛細管電泳內標法測定糖果中胭脂紅酸[J].分析測試學報,2007(1):136-138.
[9]張飛,鄭家概,付強,等.充氣糖果中胭脂紅酸的高效液相色譜測定[J].廣州化工,2013,41(24):110-111.
[10]陳永煊.UPLC-MSMS法快速檢測食品中胭脂蟲紅酸含量的研究[J].福建輕紡,2011(1):36-39.
[11]虞成華,陸志蕓,朱偉,等.反相高效液相色譜法測定食品中胭脂蟲紅色素[J].理化檢驗(化學分冊),2012,48(8):991-992.
猜你喜歡
中老年保健(2021年9期)2021-08-24 03:52:04
河北畫報(2021年2期)2021-05-25 02:07:46
中學生數理化(高中版.高考理化)(2020年2期)2020-04-21 05:33:04
兒童繪本(2020年5期)2020-04-07 17:46:30
兒童故事畫報(2019年5期)2019-05-26 14:26:14
意林原創版(2016年10期)2016-11-25 10:28:30
Coco薇(2016年2期)2016-03-22 02:42:52
山東青年(2016年1期)2016-02-28 14:25:23
Coco薇(2015年1期)2015-08-13 02:47:34
小雪花·成長指南(2015年7期)2015-08-11 15:03:12
