雙鏈RNA依賴的蛋白激酶與阿爾茨海默病相關性研究進展
2024-07-08 08:57:01龔奕肖星洋胡有生謝義煒伍知輝
中國醫學科學院學報 2024年3期
龔奕 肖星洋 胡有生 謝義煒 伍知輝
摘要:阿爾茨海默病(AD)是一種嚴重威脅人類健康的疾病,也是引起人類死亡的三大因素之一。雙鏈RNA依賴的蛋白激酶(PKR)又稱為前炎癥細胞因子,是人類先天免疫干擾素刺激因子之一。在AD發生和發展中,PKR表達上調并持續激活,一方面引發腦組織細胞發生整合應激反應,另一方面間接上調β位淀粉樣前體蛋白裂解酶1的表達,促進β淀粉樣蛋白(Aβ)的積累,而Aβ的積累又可以激活PKR,進一步促進Aβ的積累,形成一個Aβ持續積累的循環。PKR還可以促進Tau蛋白磷酸化,降低神經細胞微管穩定性。腦組織炎癥反應、Aβ的積累所引起神經毒性和微管穩定性的破壞會導致AD發生發展,引起患者記憶和認知的下降,因此PKR是AD發生發展中的關鍵分子。有效進行PKR檢測可以預測AD進展,為臨床治療AD提供先機。目前PKR已成為研發治療AD藥物的靶點,因此靶向PKR的抑制劑有望控制PKR的活性,從而有效控制AD發生發展。
關鍵詞:阿爾茨海默病;雙鏈RNA依賴的蛋白激酶;β淀粉樣蛋白;Tau蛋白
中圖分類號: R34? 文獻標識碼: A? 文章編號:1000-503X(2024)03-0425-10
DOI:10.3881/j.issn.1000-503X.15792
Research Advances in the Association Between Alzheimers Disease and Double-Stranded RNA-Dependent Protein Kinase
GONG Yi XIAO Xingyang HU Yousheng 2,XIE Yiwei3,WU Zhihui3
1Fuzhou Medical College,Nanchang University,Fuzhou,Jiangxi 344000,China
2Key Laboratory of Chronic Diseases of Fuzhou Medical College,Nanchang University,Fuzhou,Jiangxi 344000,China
3Health Science Center,Jinggangshan University,Jian,Jiangxi 343009,China
Corresponding author:HU Yousheng Tel:0794-825168 E-mail:huyousheng68@163.com
ABSTRACT:Alzheimers disease (AD) is a severe threat to human health and one of the three major causes of human death.Double-stranded RNA-dependent protein kinase (PKR) is an interferon-induced protein kinase involved in innate immunity.In the occurrence and development of AD,PKR is upregulated and continuously activated.On the one hand,the activation of PKR triggers an integrated stress response in brain cells.On the other hand,it indirectly upregulates the expression of β-site amyloid precursor protein cleaving enzyme 1 and facilitates the accumulation of amyloid-β protein (Aβ),which could activate PKR activator to further activate PKR,thus forming a sustained accumulation cycle of Aβ.In addition,PKR can promote Tau phosphorylation,thereby reducing microtubule stability in nerve cells.Inflammation in brain tissue,neurotoxicity resulted from Aβ accumulation,and disruption of microtubule stability led to the progression of AD and the declines of memory and cognitive function.Therefore,PKR is a key molecule in the development and progression of AD.Effective PKR detection can aid in the diagnosis and prediction of AD progression and provide opportunities for clinical treatment.The inhibitors targeting PKR are expected to control the activity of PKR,thereby controlling the progression of AD.Therefore,PKR could be a target for the development of therapeutic drugs for AD.
Key words:Alzheimers disease;double-stranded RNA-dependent protein kinase;amyloid-beta;Tau
Acta Acad Med Sin,2024,46(3):425-434
阿爾茨海默病(Alzheimers disease,AD)是一種慢性神經退行性病變引起的疾病。輕度AD患者首先表現為輕度認知障礙,約15%的輕度患者在兩年后病情會發展成中重度癡呆[1]。中度AD患者在記憶和語言方面會產生障礙,且難以完成洗澡和穿衣等日常事務。重度AD患者大腦中參與運動調節和控制吞咽的區域會受損,出現臥床不起、進食和飲水困難等癥狀[2]。有研究認為AD是一種復雜的疾病,由遺傳和環境因素及其他復雜的原因(如性別、年齡和不良生活習慣等)導致[3]。
AD引起老年癡呆的特征表現為思維和個人日常活動獨立性的下降,也是全球第五大死亡原因。目前全球約有5000萬AD患者,預測每5年會增長1倍,到2050年全球AD患者將可能增至1.52億。AD造成的經濟負擔不僅影響個人、家庭,同時預計還會造成全球社會經濟每年損失達1萬億美元[3]。目前尚未發現完全治愈AD的方法,現有的治療方案也只能改善患者癥狀,但AD的預防、早期診斷和早期干預卻可及時有效地延緩AD的發生發展。
目前研究者普遍認為影響AD發生發展關鍵因素是患者腦內出現的β淀粉樣蛋白(amyloid-β protein,Aβ)斑塊和Tau蛋白過度磷酸化[4-11]。研究發現,雙鏈RNA依賴的蛋白激酶(double-stranded RNA-dependent protein kinase,PKR)異常表達和激活不僅會使中樞神經系統中炎癥因子的表達增加,還會促進Aβ積累和Tau蛋白過度磷酸化,PKR在AD發生發展過程中的作用不僅體現在mRNA水平的調節上,還表現在對AD關鍵致病蛋白的翻譯調控上,因此PKR與AD的發生發展密切相關[12-18]。通過對AD患者腦組織中PKR及其作用機制的研究可以幫助我們更好地了解AD發病機制,有針對性地制訂AD的治療方案[15-18]。本文將AD與PKR關系的研究進展進行綜述。
1 AD的發病機制
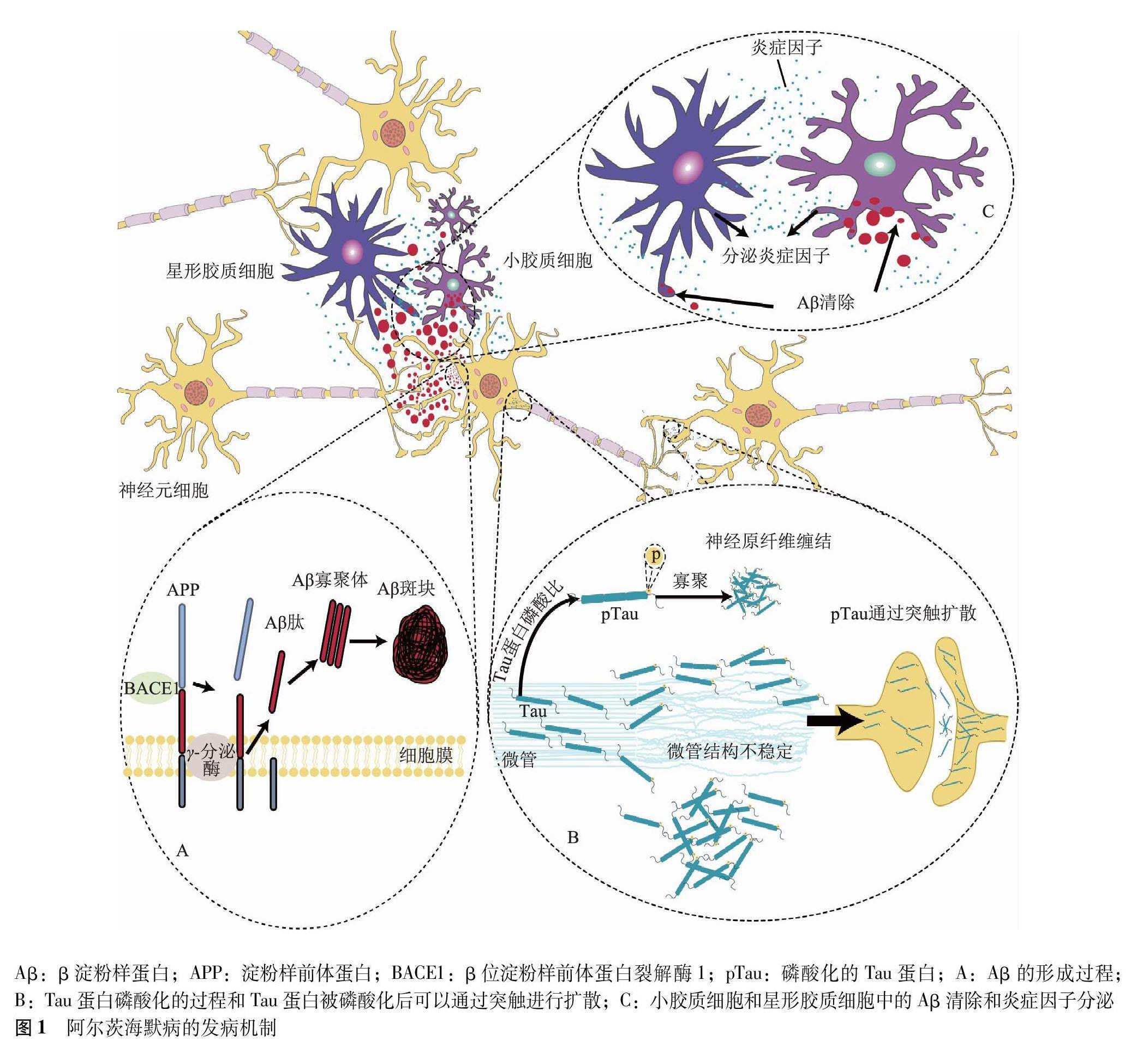
目前,研究人員普遍認為AD發病的主要機制如下:(1)Aβ積累形成的Aβ斑塊會引起炎癥等一系列反應,促使神經元細胞變性,從而誘導癡呆發生;(2)Tau蛋白變性導致神經原纖維變性,進一步引起神經元細胞變性,從而誘導癡呆發生[4]。Aβ是通過淀粉樣前體蛋白(amyloid precursor protein,APP)被β位淀粉樣前體蛋白裂解酶1(β-site amyloid precursor protein cleaving enzyme? BACE1)和γ-分泌酶依次作用后裂解釋放出Aβ[5]。Cline等[6]提出Aβ形成的級聯反應假說:Aβ及其寡聚形式的積累可能導致許多有害的后果,如出現大量炎癥反應、神經元壞死和突觸功能障礙等,從而誘導癡呆的發生。Tau蛋白又稱微管相關蛋白Tau,具有微管結合結構域,參與微管裝配和穩定,以維持細胞骨架的完整性[7-8]。Tau蛋白過度磷酸化降低其對微管的親和力,造成易形成神經原纖維纏結,并沉積在胞質溶膠中,導致不能執行維持細胞結構的功能,且這種沉積還會嚴重影響正常的細胞功能,如突觸傳遞、軸突運輸、信號轉導,導致神經細胞逐漸退化,從而誘導癡呆發生[9]。Tau蛋白磷酸化對Aβ積累會起到一定抑制作用,可能的機制是通過Tau蛋白磷酸化誘導小膠質細胞活化狀態改變,增加小膠質細胞吞噬能力,從而增強了不溶性Aβ的清除[10]。磷酸化的Tau蛋白從突觸后位點解離,成為其他激酶的底物,導致不同位點的過度磷酸化,這種過度磷酸化的Tau蛋白還可以通過軸突內連接從軸突擴散到其他健康神經元內,該過程常會引起突觸功能障礙,導致神經變性和癡呆癥狀[11](圖1)。
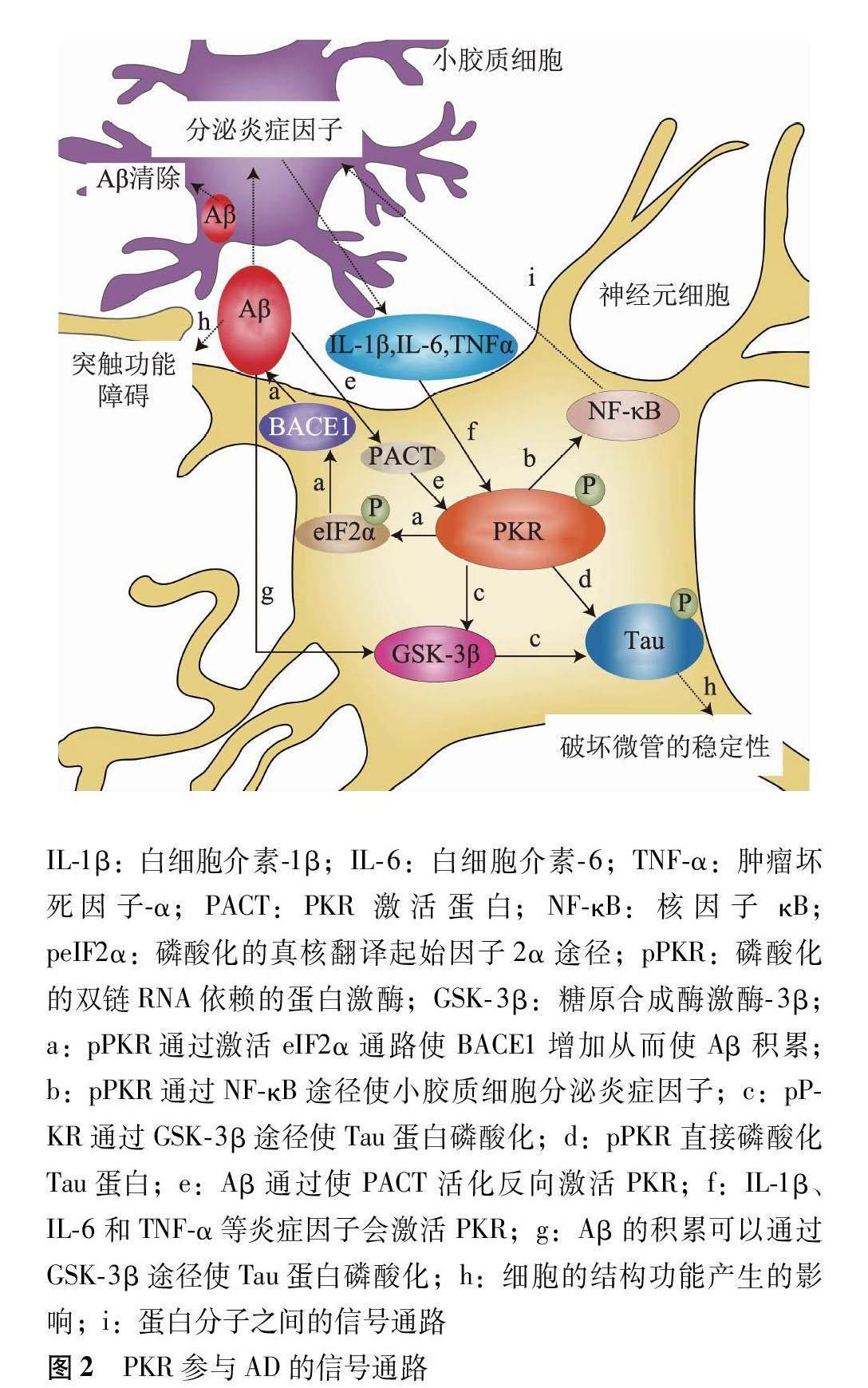
PKR是一種廣泛表達的蛋白激酶,在AD的發生發展過程中發揮了重要作用[12-18]。PKR通過多途徑參與AD的發生發展,PKR活化后能夠磷酸化真核翻譯起始因子2α(eukaryotic translation initiation factor 2α,eIF2α),從而抑制蛋白翻譯,引起神經細胞凋亡[12]。活化的PKR也可以通過核因子κB(nuclear factor κB,NF-κB)途徑激活轉錄,上調在中樞神經系統中表達的多種炎癥因子,從而引起癡呆發生[13]。PKR還可以通過控制如BACE1和γ-分泌酶等蛋白質的合成,促進Aβ積累和Tau蛋白過度磷酸化,從而影響突觸的信號傳遞和記憶認知的功能,使AD進一步惡化[14-18](圖2)。
2 PKR與AD的神經炎癥及整合應激反應
2.1 AD與整合應激反應
AD患者的主要癥狀是神經膠質細胞介導的神經炎癥。大腦中的小膠質細胞和星形膠質細胞在中樞神經系統中有著先天性免疫功能,小膠質細胞和星形膠質細胞可以介導促炎癥因子的釋放,誘導細胞發生整合應激反應,從而調節突觸產生和重組,以及還可通過非常活躍的吞噬作用和巨胞飲作用,清除死亡的神經元、細胞碎片和錯誤折疊的蛋白質,對突觸功能造成損害來影響學習和記憶[19-22]。
有研究發現,在Aβ誘導的AD模型小鼠的腦組織中,小膠質細胞和星形膠質細胞均被激活,促炎癥細胞因子表達增加,并釋放大量炎癥細胞因子;另有研究者在對死后AD樣本的組織學分析發現了腦部炎癥-老年斑周圍經常能檢測到星形膠質細胞和小膠質細胞的炎癥反應;還有研究者對AD患者腦內表達的炎癥相關基因進行統計學分析后發現,AD患者腦內的炎癥基因表達也顯著增強;這些研究結果表明炎癥可增加AD期間突觸完整性喪失和神經元變性的風險[23-27]。根據淀粉樣蛋白假說,Aβ的積累是導致AD神經退行性病變事件發生的觸發事件,在這個前提下BACE1水平升高也參與了AD的發病或進展[28-29]。當Aβ積累時,神經元凋亡并伴隨炎性細胞因子和其他蛋白表達的增加,這些
蛋白質包括白細胞介素(interleukin,IL)-1β、IL-6、IL-8和腫瘤壞死因子(tumor necrosis factor,TNF)-α等[30](圖2)。神經系統在大量的炎癥細胞因子刺激下產生慢性神經炎癥,誘導細胞發生適應性自救反應,稱為整合應激反應,該反應可以促進Aβ積累,造成神經元變性,損害突觸功能,從而影響記憶和認知能力,也是導致AD發生發展的關鍵反應之一[24-31]。
2.2 整合應激反應中的PKR及其與AD的關系
整合應激反應是指細胞在接收到應激信號后,迅速減少整體的蛋白質合成,同時增加特定蛋白翻譯以維持細胞穩態的一系列反應,該反應通過調控神經系統中重要蛋白質參與神經退行性疾病的發生發展[31]。整合應激反應的主要分子事件是激活PKR-eIF2α途徑,進而PKR又被稱為前炎癥細胞因子[31-32]。PKR在先天免疫中是一個活躍的參與者,可通過絲裂原活化蛋白激酶、干擾素調節因子3 (interferon regulatory factor 3,IRF3)和NF-κB參與炎癥反應途徑,增強炎癥反應,從而誘導AD發生發展[33]。有研究表明在AD早期,PKR可通過NF-κB和絲裂原活化蛋白激酶途徑來促進炎癥反應,誘導炎癥細胞因子釋放、增強NF-κB活性,從而增強炎癥反應[34]。
目前,有實驗用天麻素、肝豆湯改良方等制劑可以抑制PKR/eIF2α通路,從而實現了改善認知功能障礙小鼠模型中的小鼠記憶認知功能[35-36]。隨著AD患者病情進一步的發展惡化,AD晚期患者腦中的炎癥信號顯著增強,從而增加了小膠質細胞和星形膠質細胞活化,使小膠質細胞和星形膠質細胞在清除更多Aβ的同時,也通過吞噬作用清除了大量突觸,導致突觸功能減退,使大腦記憶認知功能進一步減退[24-37]。在AD晚期,Aβ又會反過來促進小膠質細胞活化,并誘導促炎癥細胞因子如IL-1β、IL-6和TNF-α等的釋放,這些炎癥因子加速神經元細胞壞死,進一步導致記憶認知功能障礙,使AD晚期患者病情加重[38-41](圖1)。
PKR對神經炎癥的調節是AD神經炎癥產生機制的重要組成部分,抑制 PKR活性可導致神經炎癥減弱,神經元的修復能力增強。有研究通過使用PKR抑制劑,發現對Aβ積累和神經炎癥均有一定的抑制作用,這一實驗支持Aβ積累會導致神經元凋亡的假設,且有研究者在動物模型中也找到Aβ積累引起神經炎性反應的證據[42]。在小膠質細胞中,抑制PKR活性也可降低Aβ引起的神經炎癥反應[42]。在最近PKR抑制劑與神經炎癥反應的相關研究中發現:(1)利福平能抑制PKR活化,減輕神經炎癥反應,從而發揮神經保護作用[43]。(2)選擇性PKR抑制劑抑制PKR活性時,AD實驗小鼠認知缺陷得到顯著改善,促炎癥細胞因子的積累和神經元變性顯著減少[42]。(3)當使用基因沉默技術降低PKR活性時,實驗小鼠的記憶和大腦的神經病理學變化表現出顯著的改善[15,18]。
3 PKR與AD的神經毒性及Aβ代謝
3.1 AD的Aβ神經毒性
Aβ的來源是APP正常切割的改變而發生的一種病理現象。APP是一種集中在神經元突觸中的單跨膜蛋白,在腦組織細胞中高度表達[44]。APP可以通過兩種不同的途徑進行加工:(1)在非淀粉樣蛋白生成途徑中,APP可以首先被α-分泌酶切割,然后被γ-分泌酶切割,產生較短的可溶性細胞外片段(非淀粉樣的);(2)在Aβ生成途徑中,APP首先被BACE1在其N-末端β域切割,而不是被α-分泌酶切割,之后被γ-C末端的分泌酶裂解,最后生成Aβ[45](圖1)。Aβ的形成過程也被稱作淀粉樣變性,是一種蛋白質的錯誤折疊,具有穩定的交叉β超二級結構、纖維狀和不溶于水等特征,也不能被蛋白水解酶降解。Aβ的積累會改變神經元細胞的正常功能,引發一系列病理事件,使神經元細胞功能障礙和死亡,最終導致突觸缺陷和記憶功能障礙[46]。Aβ早在1984年就被鑒定,其積累在AD患者腦組織的神經元細胞外,可導致AD腦組織中神經炎性斑塊的形成,神經炎性斑塊也稱為老年斑或淀粉樣斑塊,這種斑塊過度積累會嚴重影響神經元之間的信號傳遞,從而影響到患者的記憶和認知能力,導致AD發生發展[46-48]。
3.2 PKR與Aβ積累
Aβ形成的關鍵酶是BACE 由于BACE1的mRNA的5非翻譯區中含有3個上游開放閱讀框,正常情況下其表達會受到抑制,而無法翻譯出BACE1蛋白,Aβ也不會產生和積累[49]。有實驗證明BACE1的表達可被PKR的磷酸化激活,這是通過逆轉了BACE1 中mRNA的5非翻譯區對BACE1翻譯的抑制作用,從而導致了Aβ的產生與積累[18]。Tible等[18]通過蛋白質印跡法和酶聯免疫吸附測定法證實,PKR激活可解除對BACE1表達的抑制,同時激活BACE1的翻譯,進而導致Aβ的積累(圖2)。有研究者在暴露于氧化應激條件下的人神經母細胞瘤細胞中發現PKR還可以上調BACE1蛋白的水平,這表明PKR激活可以促進Aβ的產生和積累[50]。這些實驗都證實了PKR的過度活化會導致Aβ水平的增加,且與患者出現認知功能下降和記憶障礙有關[28-29,51]。
將PKR基因敲除后,AD小鼠表現出Aβ的積累水平降低,Aβ的神經毒性作用減弱,Aβ誘導的神經元細胞壞死數目明顯減少[52]。在野生型和PKR基因敲除小鼠的原代神經元中顯示,PKR的抑制劑可以明顯減少Aβ的產生和積累,這表明PKR的抑制可能對增強神經保護有效[52]。在多種實驗動物模型中,如在猴子腦室內注射Aβ寡聚體與TNF-α,可以誘導PKR和eIF2α磷酸化和認知障礙發生。在PKR和TNF-α基因敲除小鼠中,小鼠的認知功能下降和記憶障礙等癥狀可減輕,這表明TNF-α介導了PKR激活,從而誘導形成大量的Aβ寡聚體,由此所引起的神經元功能損傷和神經毒性作用[53-54]。研究者通過注射脂多糖誘導實驗小鼠產生炎癥反應和氧化應激,發現炎癥反應和氧化應激的產生,增加小鼠腦中PKR的磷酸化,而PKR的磷酸化又會使Aβ的產生和積累,引起記憶與運動功能的障礙。通過對實驗小鼠進行PKR基因的敲除/抑制后,Aβ水平會降低,記憶與運動功能的障礙也可改善[33,55-56]。
有研究通過對AD患者、APP/PS1 KI轉基因小鼠和暴露于氧化應激的培養細胞進行免疫熒光檢測來評估PKR激活蛋白(PKR associated protein activator,PACT)和PKR的表達水平,實驗結果表明,AD患者的大腦中Aβ可誘導PACT的表達,使PKR激活,這說明Aβ的積累會反向激活PKR[57]。理論上,減少Aβ可減少PKR激活,所以通過抑制PKR激活,同時聯合使用藥物降低Aβ可以用來治療AD。
總的來說,PKR在被炎癥刺激的神經元細胞中表達增加,PKR激活后可通過誘導BACE1表達來上調Aβ的表達,反過來Aβ的積累可以通過活化PACT來激活PKR,又進一步促進Aβ的積累,形成了一個惡性循環,使AD進一步發展惡化(圖2)。
4 PKR與AD突觸傳導及Tau蛋白磷酸化
4.1 Tau蛋白磷酸化對AD突觸傳導的影響
Tau蛋白由1個微管相關蛋白Tau基因編碼,存在6種亞型[58]。Tau蛋白是一種主要在神經元軸突中表達的微管相關蛋白,其主要功能是促進微管的組裝和穩定性[59]。在AD和其他Tau蛋白病的發生發展過程中,Tau蛋白的過度磷酸化是最早發生和持續病變事件之一[60]。磷酸化Tau蛋白可破壞突觸組成和結構,導致突觸功能障礙或突觸功能的完全喪失[1 61]。當磷酸化Tau蛋白不斷聚集后,可通過突觸向未出現磷酸化Tau的正常神經元擴散,進一步使AD的病情惡化[62-63]。
4.2 PKR與Tau蛋白磷酸化
PKR參與了Tau蛋白的磷酸化,在對AD患者腦組織切片的檢測中證實,磷酸化的PKR可以與磷酸化的Tau蛋白在受影響的神經元中共定位[64-65]。最近研究發現,PKR過表達會增加HEK-293T細胞中Tau蛋白及其mRNA的水平[62]。PKR沉默可降低SH-SY5Y細胞中Tau蛋白及其mRNA水平,在腦組織急性炎癥中PKR可以不依賴其他激酶直接磷酸化Tau蛋白的多個和疾病相關的殘基,觸發Tau蛋白的病理轉變,這說明PKR可以直接調控Tau蛋白的表達和Tau蛋白的磷酸化[62,66]。
PKR的激活與磷酸化的Tau蛋白聚集在AD病理發展中也扮演了重要角色,研究者通過對具有AD標志病變的淀粉樣蛋白斑塊和Tau磷酸化病變的人類神經元細胞的研究發現:磷酸化Aβ積累會使糖原合成酶激酶3β激活并導致磷酸化的Tau蛋白聚集,而PKR磷酸化又可以通過激活糖原合成酶激酶3β間接導致磷酸化的Tau蛋白聚集[67](圖2)。
有研究提出在 AD發病早期,磷酸化的Tau蛋白聚集會嚴重影響微管細胞骨架的穩定性,從而使AD病情進一步惡化發展,PKR磷酸化直接和間接誘導Tau蛋白磷酸化,磷酸化Tau蛋白從微管中脫落,破壞了微管細胞骨架的穩定,從而使軸突的信號傳導障礙,導致AD患者出現記憶認知功能的障礙,因為PKR激活會使Tau蛋白磷酸化,同時促進Tau蛋白從微管中移除的病理轉變,所以PKR連接了AD神經變性中的Aβ積累機制和Tau蛋白磷酸化機制[62](圖2)。
5 PKR與AD的診斷和臨床治療
5.1 PKR與AD的診斷
到目前為止,很難在血液或腦脊液中找到一種可靠的生物標志物來預測AD患者的認知衰退程度。AD患者大腦中活化的PKR水平有所增加,且AD患者的外周血淋巴細胞中的總PKR及磷酸化PKR水平均有所升高[68-69]。研究者通過對PKR進行抑制后來觀察對患者的記憶、認知等的影響,發現當抑制PKR之后,患者明顯減少了神經元的損傷和變性,Tau蛋白的磷酸化水平也降低,同時恢復了突觸可塑性,又減少神經炎癥等,因此PKR有希望成為未來預測AD的標志物[18,35,43,63]。
大量科研團隊開始進行PKR與AD疾病預測及后續發展的趨勢相關性的研究。有研究發現通過檢測腦脊液中磷酸化PKR濃度可以預測AD患者未來的認知功能衰退程度[70]。有研究證明了磷酸化PKR較經典生物標志物對AD患者病情發展的速度預測有更好的效果,但是腦脊液抽取存在侵入性、有創性以及成像技術的高昂費用等問題,研究者希望還是尋找血源性生物標志物進行AD預測和診斷[71]。生曉娜等[68]通過臨床研究發現AD 患者的外周血淋巴細胞中總PKR及磷酸化PKR的水平均較正常組有明顯升高,同時對認知水平和PKR水平之間進行統計學分析發現AD患者淋巴細胞中的總PKR與磷酸化PKR水平與認知障礙嚴重程度呈正相關。Monllor等[69]通過研究證明PKR與Aβ積累相關,PKR的血清濃度反映了患者的淀粉樣病變程度,說明PKR是AD的潛在生物標志物。這些研究結果表明使用PKR檢測比傳統的檢測方法更有助于助于AD的早期診斷,同時還可以更好地檢測AD的發展和預后情況,PKR檢測在未來或許可以成為 AD病情的早期診斷及評價的一種更有效的方法。
5.2 PKR對AD治療的潛在作用
AD通常以記憶和認知能力下降為標志,PKR的激活與記憶和認知功能障礙有關,PKR的上調和激活會加速細胞外Aβ形成,導致大腦海馬區神經元細胞凋亡增加,PKR的激活可以通過促進Tau蛋白磷酸化而抑制神經突觸的形成和發展,因此PKR是影響記憶和認知過程中的關鍵分子[15,62,72]。
天麻素在AD轉基因Tg2576小鼠中作為PKR抑制劑,能降低AD實驗小鼠發生記憶障礙的水平[36]。在AD轉基因模型5xFAD小鼠中PKR基因缺失可以減少5xFAD小鼠的發生記憶障礙,且部分挽救了在5xFAD小鼠模型中觀察到的空間記憶缺陷[18]。這些實驗均反映出PKR對AD的發展和惡化起重要作用,為未來通過抑制PKR來治療或緩解AD提供了思路。
目前已有研究開始探索PKR在AD發展中的作用的臨床應用,但還未開發出有效臨床治療藥物[73]。默克公司研發的治療AD候選藥物verubecestat完成了兩項大型Ⅲ期臨床試驗,一項針對輕度至中度AD,另一項針對前驅期AD,但這兩項試驗均因無效而提前終止,其中失敗的因素還需要通過進一步的試驗來探索論證[73-74]。
6 總結與展望
綜上,在AD進展過程中,PKR起著關鍵的作用。PKR活化可通過一個閉環反復活化,導致Aβ不斷積累。PKR活化可以通過NF-κB途徑促進炎癥細胞因子的表達,還可以通過PKR-eIF2α途徑抑制蛋白翻譯,引起神經細胞凋亡,從而使AD進一步發展。
AD是一個多因素引發、發病機制復雜的疾病。目前,AD尚無有效檢測預測和治療手段。PKR參與了AD發生發展的多個病理生理過程,特別是在PKR活化eIF2α磷酸化,整體蛋白翻譯抑制的條件下,APP和炎癥細胞因子蛋白卻表達上調,并在AD的發展中導致Aβ積累和炎癥反應增強。因此,對于AD進展過程中,蛋白翻譯表達調控機制仍需要深入地研究,AD患者中PKR表達譜、突變譜和PKR在AD進程中的活性變化也需要進一步研究。AD診療需開發更有效、準確地檢測PKR來預測AD發生發展的試劑盒,并進一步開展PKR抑制劑研究,開發出有效緩解和治療AD的相關藥物。
利益沖突 所有作者聲明無利益沖突
作者貢獻聲明 龔奕:文獻研究、圖片繪制、文稿撰寫和修改;肖星洋:文獻篩選和整理分類;胡有生:研究思路和撰寫大綱提出;謝義煒:流行病學和臨床醫學數據搜集和分析;伍知輝:參與文稿撰寫和校正,并同意対研究工作誠信負責
參 考 文 獻
[1]Petersen RC,Lopez O,Armstrong MJ,et al.Practice guideline update summary:mild cognitive impairment:report of the Guideline Development,Dissemination,and Implementation Subcommittee of the American Academy of Neurology[J].Neurology,2018,90(3):126-135.DOI:10.1212/wnl.0000000000004826.
[2]Alzheimers Association.2023 Alzheimers disease facts and figures[J].Alzheimers Dement,2023,19(4):1598-1695.DOI:10.1002/alz.13016.
[3]Livingston G,Huntley J,Sommerlad A,et al.Dementia prevention,intervention,and care:2020 report of the Lancet Commission[J].Lancet (London,England),2020,396(10248):413-446.DOI:10.1016/s0140-6736(20)30367-6.
[4]Kapasi A,Leurgans SE,Arvanitakis Z,et al.Abeta (Amyloid Beta) and Tau tangle pathology modifies the association between small vessel disease and cortical microinfarcts[J].Stroke,202 52(3):1012-1021.DOI:10.1161/STROKEAHA.120.031073.
[5]Behl T,Kaur I,Fratila O,et al.Exploring the potential of therapeutic agents targeted towards mitigating the events associated with Amyloid-β cascade in Alzheimers disease[J].Int J Mol Sci,2020,21(20):7473.DOI:10.3390/ijms21207443.
[6]Cline EN,Bicca MA,Viola KL,et al.The amyloid-β oligomer hypothesis:beginning of the third decade[J].J Alzheimers Dis,2018,64(s1):S567-S610.DOI:10.3233/jad-179941.
[7]Hamano T,Enomoto S,Shirafuji N,et al.Autophagy and Tau protein[J].Int J Mol Sci,202 22(14):7574.DOI:10.3390/ijms22147475.
[8]Otero-Garcia M,Mahajani SU,Wakhloo D,et al.Molecular signatures underlying neurofibrillary tangle susceptibility in Alzheimers disease[J].Neuron,2022,110(18):2929-2948.e8.DOI:10.1016/j.neuron.2022.06.021.
[9]Huang F,Wang M,Liu R,et al.CDT2-controlled cell cycle reentry regulates the pathogenesis of Alzheimers disease[J].Alzheimers Dement,2019,15(2):217-231.DOI:10.1016/j.jalz.2018.08.013.
[10]Wang Q,Xie C.Microglia activation linking amyloid-β drive tau spatial propagation in Alzheimers disease[J].Front Neurosci,2022,16:951128.DOI:10.3389/fnins.2022.951128.
[11]Ittner A,Ittner LM.Dendritic Tau in Alzheimers disease[J].Neuron,2018,99(1):13-27.DOI:10.1016/j.neuron.2018.06.003.
[12]Chukwurah E,Farabaugh KT,Guan BJ,et al.A tale of two proteins:PACT and PKR and their roles in inflammation[J].FEBS J,202 288(22):6365-6391.DOI:10.1111/febs.15691.
[13]Lee S,Jee HY,Lee YG,et al.PKR-Mediated phosphorylation of eIF2a and CHK1 is associated with doxorubicin-mediated apoptosis in hCC1143 triple-negative breast cancer cells[J].Int J Mol Sci,2022,23(24):15872.DOI:10.3390/ijms232415872.
[14]Zhang J,Zhang X,Li L,et al.Activation of double-stranded RNA-activated protein kinase in the dorsal root ganglia and spinal dorsal horn regulates neuropathic pain following peripheral nerve injury in rats[J].Neurotherapeutics,2022,19(4):1381-1400.DOI:10.1007/s13311-022-01255-2.
[15]Lu W,Tang S,Li A,et al.The role of PKC/PKR in aging,Alzheimers disease,and perioperative neurocognitive disorders[J].Front Aging Neurosci,2022,14:973068.DOI:10.3389/fnagi.2022.973068.
[16]Upadhyay A,Chhangani D,Rao NR,et al.Amyloid fibril proteomics of AD brains reveals modifiers of aggregation and toxicity[J].Mol Neurodegener,2023,18(1):61.DOI:10.1186/s13024-023-00654-z.
[17]Futamura A,Hieda S,Mori Y,et al.Toxic Amyloid-β42 conformer may accelerate the onset of Alzheimers disease in the preclinical stage[J].J Alzheimers Dis,202 80(2):639-646.DOI:10.3233/jad-201407.
[18]Tible M,Mouton Liger F,Schmitt J,et al.PKR knockout in the 5xFAD model of Alzheimers disease reveals beneficial effects on spatial memory and brain lesions[J].Aging cell,2019,18(3):e12887.DOI:10.1111/acel.12887.
[19]Hasan U,Singh SK.The Astrocyte-Neuron Interface:An overview on molecular and cellular dynamics controlling formation and maintenance of the tripartite synapse[J].Methods Mol Biol,2019,1938:3-18.DOI:10.1007/978-1-4939-9068-9_1.
[20]Ikegami A,Haruwaka K,Wake H.Microglia:lifelong modulator of neural circuits[J].Neuropathology,2019,39(3):173-180.DOI:10.1111/neup.12560.
[21]Konishi H,Kiyama H,Ueno M.Dual functions of microglia in the formation and refinement of neural circuits during development[J].Int J Dev Neurosci,2019,77:18-25.DOI:10.1016/j.ijdevneu.2018.09.009.
[22]Olsen M,Aguilar X,Sehlin D,et al.Astroglial responses to amyloid-beta progression in a mouse model of Alzheimers disease[J].Mol Imaging Biol,2018,20(4):605-614.DOI:10.1007/s11307-017-1153-z.
[23]Litwiniuk A,Juszczak GR,Stankiewicz AM,et al.The role of glial autophagy in Alzheimers disease[J].Mol Psychiatry,2023,28(11):4528-4539.DOI:10.1038/s41380-023-02242-5.
[24]Liu Q,Contreras A,Afaq MS,et al.Intensity-dependent gamma electrical stimulation regulates microglial activation,reduces beta-amyloid load,and facilitates memory in a mouse model of Alzheimers disease[J].Cell Biosci,2023,13(1):138.DOI:10.1186/s13578-023-01085-5.
[25]Zhang Y,Jia J.Betaine Mitigates Amyloid-β-Associated neuroinflammation by suppressing the NLRP3 and NF-κB signaling pathways in microglial cells[J].J Alzheimers Dis,2023,94(s1):S9-S19.DOI:10.3233/jad-230064.
[26]Liu W,Chen S,Rao X,et al.The inflammatory gene PYCARD of the entorhinal cortex as an early diagnostic target for Alzheimers disease[J].Biomedicines,2023,11(1):194.DOI:10.3390/biomedicines11010194.
[27]panic′ E,Langer Horvat L,Ilic′ K,et al.NLRP1 inflammasome activation in the hippocampal formation in Alzheimers disease:correlation with neuropathological changes and unbiasedly estimated neuronal loss[J].Cells,2022,11(14):2223.DOI:10.3390/cells11142223.
[28]Bouteiller JC,Mergenthal AR,Hu E,et al.Pathogenic processes underlying Alzheimers disease:modeling the effects of Amyloid beta on synaptic transmission[J].Annu Int Conf IEEE Eng Med Biol Soc,2019,2019:1956-1959.DOI:10.1109/embc.2019.8857871.
[29]Castellani RJ,PLASCENCIA-VILLA G,Perry G.The amyloid cascade and Alzheimers disease therapeutics:theory versus observation[J].Lab Invest,2019,99(7):958-970.DOI:10.1038/s41374-019-0231-z.
[30]Ratan Y,Rajput A,Maleysm S,et al.An insight into cellular and molecular mechanisms underlying the pathogenesis of neurodegeneration in Alzheimers disease[J].Biomedicines,2023,11(5):1398.DOI:10.3390/biomedicines11051398.
[31]Bond S,Lopez-Lloreda C,Gannon PJ,et al.The integrated stress response and phosphorylated eukaryotic initiation factor 2α in neurodegeneration[J].J Neuropathol Exp Neurol,2020,79(2):123-143.DOI:10.1093/jnen/nlz129.
[32]Qiao H,Jiang T,Mu P,et al.Cell fate determined by the activation balance between PKR and SPHK1[J].Cell Death Differ,202 28(1):401-418.DOI:10.1038/s41418-020-00608-8.
[33]Hugon J,Paquet C.The PKR/P38/RIPK1 signaling pathway as a therapeutic target in Alzheimers disease[J].Int J Mol Sci,202 22(6):3136.DOI:10.3390/ijms22063136.
[34]Chiarini A,Armato U,Hu P,et al.Danger-Sensing/Patten recognition receptors and neuroinflammation in Alzheimers disease[J].Int J Mol Sci,2020,21(23):9036.DOI:10.3390/ijms21239036.
[35]劉松楊,程楠,徐陳陳,等.肝豆湯改良方調控PKR/eIF2α通路改善Wilson病模型TX小鼠突觸功能障礙的機制研究[J].安徽中醫藥大學學報,202 40(6):75-81.DOI:10.3969/j.issn.2095-7246.2021.06.017.
[36]Zhang J S,Zhou SF,Wang Q,et al.Gastrodin suppresses BACE1 expression under oxidative stress condition via inhibition of the PKR/eIF2α pathway in Alzheimers disease[J].Neuroscience,2016,325:1-9.DOI:10.1016/j.neuroscience.2016.03.024.
[37]Ries M,Sastre M.Mechanisms of Aβ clearance and degradation by glial cells[J].Front Aging Neurosci,2016,8:160.DOI:10.3389/fnagi.2016.00160.
[38]Khandelwal PJ,Herman AM,Moussa CE.Inflammation in the early stages of neurodegenerative pathology[J].J Neuroimmunol,201 238(1-2):1-11.DOI:10.1016/j.jneuroim.2011.07.002.
[39]Deng Z,Dong Y,Zhou X,et al.Pharmacological modulation of autophagy for Alzheimers disease therapy:opportunities and obstacles[J].Acta Pharm Sin B,2022,12(4):1688-1706.DOI:10.1016/j.apsb.2021.12.009.
[40]Suresh S,Larson J,Jenrow KA.Chronic neuroinflammation impairs waste clearance in the rat brain[J].Front Neuroanat,2022,16:1013808.DOI:10.3389/fnana.2022.1013808.
[41]Doroszkiewicz J,Mroczko P,Kulczyńska-Przybik A.Inflammation in the CNS:understanding various aspects of the pathogenesis of Alzheimers disease[J].Curr Alzheimer Res,2022,19(1):16-31.DOI:10.2174/1567205018666211202143935.
[42]Lopez-Grancha M,Bernardelli P,Moindrot N,et al.A novel selective PKR inhibitor restores cognitive deficits and neurodegeneration in Alzheimer disease experimental models[J].J Pharmacol Exp Ther,202 378(3):262-275.DOI:10.1124/jpet.121.000590.
[43]鄧嘉強,井秀娜,林淡鈺,等.利福平通過抑制蛋白激酶R活化調節魚藤酮誘導的小膠質細胞炎癥而發揮神經保護作用[J].嶺南急診醫學雜志,202 26(2):121-124.DOI:10.3969/j.issn.1671-301X.2021.02.004.
[44]Rice HC,de Malmazet D,Schreurs A,et al.Secreted amyloid-β precursor protein functions as a GABA(B)R1a ligand to modulate synaptic transmission[J].Science,2019,363(6423):eaao4827.DOI:10.1126/science.aao4827.
[45]Maucat-Tan NB,Saadipour K,Wang YJ,et al.Cellular trafficking of amyloid precursor protein in amyloidogenesis physiological and pathological significance[J].Mol Neurobiol,2019,56(2):812-830.DOI:10.1007/s12035-018-1106-9.
[46]Ma C,Hong F,Yang S.Amyloidosis in Alzheimers disease:pathogeny,etiology,and related therapeutic directions[J].Molecules,2022,27(4):1210.DOI:10.3390/molecules27041210.
[47]Glenner GG,Wong CW.Alzheimers disease:initial report of the purification and characterization of a novel cerebrovascular amyloid protein[J].Biochem Biophys Res Commun,1984,120(3):885-890.DOI:10.1016/s0006-291x(84)80190-4.
[48]Yakupova EI,Bobyleva LG,Shumeyko SA,et al.Amyloids:the history of toxicity and functionality[J].Biology,202 10(5):394.DOI:10.3390/biology10050394.
[49]Guix FX,Sartório CL,Ill-Raga G.BACE1 translation:at the crossroads between Alzheimers disease neurodegeneration and memory consolidation[J].J Alzheimers Dis Rep,2019,3(1):113-148.DOI:10.3233/adr-180089.
[50]Syeda T,Cannon JR.Environmental exposures and the etiopathogenesis of Alzheimers disease:the potential role of BACE1 as a critical neurotoxic target[J].J Biochem Mol Toxicol,202 35(4):e22694.DOI:10.1002/jbt.22694.
[51]Volloch V,Rits-Volloch S.The amyloid cascade hypothesis 2.0 for Alzheimers disease and aging-associated cognitive decline:from molecular basis to effective therapy[J].Int J Mol Sci,2023,24(15):12246.DOI:10.3390/ijms241512246.
[52]Gourmaud S,Mouton-Liger F,Abadie C,et al.Dual kinase inhibition affords extended in vitro neuroprotection in Amyloid-β toxicity[J].J Alzheimers Dis,2016,54(4):1659-1670.DOI:10.3233/jad-160509.
[53]Lourenco MV,Clarke JR,Frozza RL,et al.TNF-α mediates PKR-dependent memory impairment and brain IRS-1 inhibition induced by Alzheimers β-amyloid oligomers in mice and monkeys[J].Cell Metab,2013,18(6):831-843.DOI:10.1016/j.cmet.2013.11.002.
[54]Chan AWS,Cho IK,Li CX,et al.Cerebral Aβ deposition in an Aβ-precursor protein-transgenic rhesus monkey[J].Aging Brain,2022,2:100044.DOI:10.1016/j.nbas.2022.100044.
[55]Mouton-Liger F,Rebillat AS,Gourmaud S,et al.PKR downregulation prevents neurodegeneration and β-amyloid production in a thiamine-deficient model[J].Cell Death Dis,2015,6(1):e1594.DOI:10.1038/cddis.2014.552.
[56]Carret-Rebillat AS,Pace C,GourmauDS,et al.Neuroinflammation and Aβ accumulation linked to systemic inflammation are decreased by genetic PKR down-regulation[J].Sci Rep,2015,5:8489.DOI:10.1038/srep08489.
[57]Paquet C,Mouton-Liger F,Meurs EF,et al.The PKR activator PACT is induced by Aβ:involvement in Alzheimers disease[J].Brain Pathol,2012,22(2):219-229.DOI:10.1111/j.1750-3639.2011.00520.x.
[58]Chen Y,Yu Y.Tau and neuroinflammation in Alzheimers disease:interplay mechanisms and clinical translation[J].J Neuroinflammation,2023,20(1):165.DOI:10.1186/s12974-023-02853-3.
[59]el Mammeri N,Duan P,Dregni AJ,et al.Amyloid fibril structures of tau:conformational plasticity of the second microtubule-binding repeat[J].Sci Adv,2023,9(28):eadh4731.DOI:10.1126/sciadv.adh4731.
[60]Congdon EE,Sigurdsson EM.Tau-targeting therapies for Alzheimer disease[J].Nat Rev Neurol,2018,14(7):399-415.DOI:10.1038/s41582-018-0013-z.
[61]Dejanovic B,Huntley MA,de Mazière A,et al.Changes in the synaptic proteome in tauopathy and rescue of Tau-induced synapse loss by c1q antibodies[J].Neuron,2018,100(6):1322-1336.e7.DOI:10.1016/j.neuron.2018.10.014.
[62]Reimer L,Betzer C,Kofoed RH,et al.PKR kinase directly regulates tau expression and Alzheimers disease-related tau phosphorylation[J].Brain Pathol,202 31(1):103-119.DOI:10.1111/bpa.12883.
[63]Dubbelman MA,Mimmack KJ,Sprague EH,et al.Regional cerebral tau predicts decline in everyday functioning across the Alzheimers disease spectrum[J].Alzheimers Res Ther,2023,15(1):120.DOI:10.1186/s13195-023-01267-w.
[64]Moradi Majd R,Mayeli M,Rahmani F.Pathogenesis and promising therapeutics of Alzheimer disease through eIF2α pathway and correspondent kinases[J].Metab Brain Dis,2020,35(8):1241-1250.DOI:10.1007/s11011-020-00600-8.
[65]Drummond E,Pires G,Macmurray C,et al.Phosphorylated tau interactome in the human Alzheimers disease brain[J].Brain,2020,143(9):2803-2817.DOI:10.1093/brain/awaa223.
[66]Turab Naqvi AA,Hasan GM,Hassan MI.Targeting Tau hyperphosphorylation via kinase inhibition:strategy to address Alzheimers disease[J].Curr Top Med Chem,2020,20(12):1059-1073.DOI:10.2174/1568026620666200106125910.
[67]Amin J,Paquet C,Baker A,et al.Effect of amyloid-β (Aβ) immunization on hyperphosphorylated tau:a potential role for glycogen synthase kinase (GSK)-3β[J].Neuropathol Appl Neurobiol,2015,41(4):445-457.DOI:10.1111/nan.12205.
[68]生曉娜,李瀟瀟,張曉煒,等.阿爾茨海默病患者淋巴細胞中雙鏈RNA-依賴的蛋白激酶水平與認知障礙的相關性[J].中風與神經疾病雜志,2017,34(8):692-695.DOI:10.19845/j.cnki.zfysjjbzz.2017.08.005.
[69]Monllor P,Giraldo E,Badia MC,et al.Serum levels of clusterin,PKR,and RAGE correlate with amyloid burden in Alzheimers disease[J].J Alzheimers Dis,202 80(3):1067-1077.DOI:10.3233/jad-201443.
[70]Dumurgier J,Mouton-Liger F,Lapalus P,et al.Cerebrospinal fluid PKR level predicts cognitive decline in Alzheimers disease[J].PLoS One,2013,8(1):e53587.DOI:10.1371/journal.pone.0053587.
[71]Paquet C,Dumurgier J,Hugon J.Pro-apoptotic kinase levels in cerebrospinal fluid as potential future biomarkers in Alzheimers disease[J].Front Neurol,2015,6:168.DOI:10.3389/fneur.2015.00168.
[72]Zeng Y,Wang L,Zhou Y,et al.NMDA receptor antagonists engender neuroprotection against gp120-induced cognitive dysfunction in rats through modulation of PKR activation,oxidative stress,ER stress and IRE1α signal pathway[J].Eur J Neurosci,2022,56(2):3806-3824.DOI:10.1111/ejn.15688.
[73]Egan MF,Kost J,Voss T,et al.Randomized trial of verubecestat for prodromal Alzheimers disease[J].N Engl J Med,2019,380(15):1408-1420.DOI:10.1056/NEJMoa1812840.
[74]Egan MF,Kost J,Tariot PN,et al.Randomized trial of verubecestat for mild-to-moderate Alzheimers disease[J].N Engl J Med,2018,378(18):1691-1703.DOI:10.1056/NEJMoa1706441.
(收稿日期:2023-08-08)
猜你喜歡
興趣閱讀·興趣作文與閱讀(低年級)(2025年8期)2025-08-18 00:00:00
學苑創造·A版(2020年10期)2020-11-06 05:21:26
學苑創造·A版(2020年9期)2020-10-13 09:41:02
小學生學習指導(低年級)(2017年10期)2017-10-10 01:00:05
作文周刊·小學一年級版(2016年27期)2017-06-03 23:21:17
絲綢之路(2016年9期)2016-05-14 14:36:33
新湘評論·下半月(2016年4期)2016-05-05 22:12:41
新湘評論·下半月(2016年4期)2016-05-05 22:12:41
海外文摘(2016年4期)2016-04-15 22:28:55
云南中醫學院學報(2014年3期)2014-07-31 18:57:34
