藥品質量標準中雜質的限度確定方式探討
2024-10-12 00:00:00闞洪峰宋錫瑞
品牌與標準化 2024年5期
【摘要】本文將對創新藥品質量標準中雜質的限度確定方式進行探討,旨在尋找最優化的雜質限制策略,以提高藥品質量,保障患者用藥安全和公眾健康。在這個過程中,本文將分析不同類型的藥品雜質、它們的來源以及對藥物療效和安全性的影響,同時,還將探討現有的雜質限定方法及其局限性,針對這些問題,提出更科學、合理的雜質限度確定方式和改進策略。
【關鍵詞】藥品質量標準;雜質;限度確定;方式探討
【DOI編碼】10.3969/j.issn.1674-4977.2024.05.039
Discussion on the Determination of Impurity Limits in Drug Quality Standards
KAN Hongfeng, SONG Xirui*
(Qilu Pharmaceutical Co., Ltd., Jinan 250100, China)
Abstract: This article will explore the method of determining the limit of impurities in innovative drug quality standards, aiming to find the optimal impurity restriction strategy to improve drug quality, ensure patient medication safety and public health. In this process, different types of drug impurities, their sources, and their impact on drug efficacy and safety will be analyzed. At the same time, existing impurity limitation methods and their limitations will be explored. To address these issues, more scientific and reasonable impurity limit determination methods and improvement strategies will be proposed.
Keywords: drug quality standards; impurities; limit determination; exploration of methods
0引言
隨著科學技術的迅速發展和醫藥行業的不斷進步,藥品的質量控制越來越受到重視,藥品質量的關鍵因素之一便是其中的雜質含量,其會直接影響藥品的療效、安全性和穩定性,制定合理的藥品雜質限度標準對于保障藥品質量至關重要。因此,需要對藥品質量標準中雜質的限度確定方式進行分析,從而保證藥品質量。
1藥品質量標準中雜質的限度概述
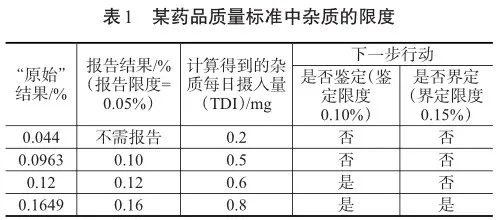
藥品質量標準中雜質的限度是指在藥品成分中可以接受的雜質含量上限,這些雜質在藥物的生產、儲存和使用過程中不可避免地產生,這些雜質通常包括有機雜質、無機雜質和殘留溶劑等。例如,有機雜質可能包括來自原料藥的前體、副產物、降解產物等;無機雜質可能包括重金屬、非金屬元素和催化劑等;殘留溶劑則是藥物生產過程中未能完全去除的溶劑。對于這些雜質,國際藥典委員會等權威組織制定了嚴格的限度標準,以保證藥品的療效和安全性。通常情況下,這些限度標準是根據雜質毒性臨床數據、藥物暴露度和藥品特性等因素確定的。在確定藥品雜質限度時,通常采用的方法有定性分析和定量分析。定性分析主要采用如高效液相色譜法(HPLC)、氣相色譜法(GC)和紫外分光光度法(UV)等技術來鑒定雜質種類;定量分析則采用如熒光法、原子吸收光譜法(AAS)或電感耦合等離子質譜法(ICP-MS)等技術來測定雜質含量。然而,無論采用哪種技術,都應注重方法的選擇、建立和驗證。例如,在建立方法時,需要評估線性范圍、檢測限、定量限、精密度和準確度等參數,以便科學地解決實際問題,并使測量結果具有可靠性和穩定性。關于藥品雜質限度標準的確定,現行藥典和相關指導文件提供了可操作性強、科學性較高的指南。然而,限度標準的設置仍存在不足之處,如缺乏統一標準、難以適用于所有類型藥物等。因此,未來發展方向應注重個性化限度策略,根據不同藥物的特性、患者人群和臨床狀況等因素,制定靈活、可靠的藥品雜質限度標準[1]。為更好地保障藥品的療效、安全性和穩定性,有關部門和研究人員應不斷更新監管政策、完善檢測方法和提高行業標準,以便進一步優化藥品質量管理體系。某藥品質量標準中雜質的限度見表1。
2不同類型的藥品雜質及來源
2.1雜質源于合成過程中的反應副產物
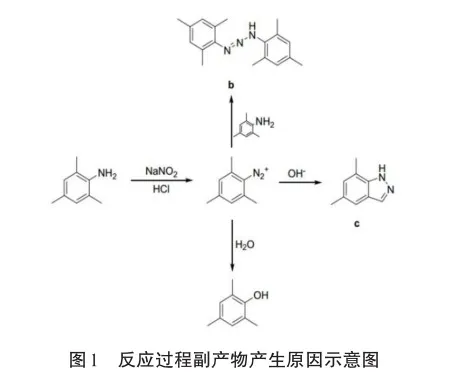
合成類藥物在生產過程中,往往會產生反應副產物,導致藥物雜質的產生。實際生產過程中,許多化學反應產物的分離程度很難達到100%的純度。如苯甲酸酯類藥物,包括布洛芬、消炎痛等,容易在不完全溶解或部分水解等條件下形成苯甲酸異構體為雜質。EPA(美國環保署)限量規定的重量百分比(w/w)含量,基于含量95%~101%的藥物,可允許某些雜質含量達到0.3%。有關藥品雜質的偏差應該符合ICH-Q3A(International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use)和ICH-Q3B政策中關于實際制藥期間特定化合物的最高允許濃度的相關要求。為了進一步減少雜質的生成,優化合成條件,如對反應條件(反應時間、溫度、溶劑、催化劑選擇等)的嚴格控制和藥物制程管理,有助于降低雜質產生的概率。反應過程副產物產生原因示意圖見圖1。
2.2雜質來自合成藥物中的起始材料、中間體、試劑和催化劑
藥物合成過程中采用的起始材料、中間體、試劑和催化劑一般具有復雜化學結構,未充分反應或反應過程中產生的副產物可能成為藥品中的雜質。例如,在苗頭類藥物羅華托品(Rosuvastatin)的合成過程中,使用的N-甲基-2-吡啶甲醇(NMP)郁金香用作反應劑,在反應過程中可能形成其酰胺類結構,作為雜質存在。為了減少這類雜質,需關注材料質量、藥品合成過程中的分級反應優化并降低剩余試劑/催化劑的含量。生產過程中遵循GMP(Good Manufacturing Practice)原則,并根據藥品生產中的關鍵工藝參數(如進料純度等)確保藥物質量。
2.3降解雜質
藥品雜質常見于各類藥物中,這些雜質來源于制藥過程、貯存條件以及藥物本身的化學結構。藥品雜質分為有機雜質、無機雜質和水合物。有機雜質包括原料藥、溶劑殘留、雜原子及未參與反應的官能團。例如,含氯代烷基的藥品在溶劑回收后可能存在0.1~3 ppm的氯甲烷殘留。無機雜質通常來源于制藥過程中的催化劑、重金屬、鹽化劑等,如鈀或銠在苯甲酸華法林中的殘留量需控制在50 ppb以下。水合物則包括藥品分子與水分子結合形成的雜質,如阿莫西林三水合物的水分為11.5%~12.5%。降解雜質是藥品在貯存、加工等過程中形成的一種雜質,這些雜質受制藥工藝、溶劑、反應條件、貯存條件、光照、濕度和pH值等因素影響。
3藥品質量標準中雜質的限度確定方式
3.1雜質生物毒性和藥理活性試驗
評估限量方法的安全性主要依賴于對藥品雜質的生物毒性和藥理活性的認識。首先,可以通過比較雜質與目標物質在分子結構上的相似性,初步推測它們可能具有的藥理活性和生物毒性特征。然后,針對某一特定雜質展開更為詳細的實驗室研究,如動物實驗(包括小鼠、大鼠、猴子等模型)、體外細胞培養以及生物分析等,并分析相關的藥代動力學參數,如半衰期(t1/2)、暴露總量和最大血藥濃度。在藥品雜質實驗中,通過實驗所獲得的每個雜質的無觀察毒性反應劑量(NOEL)、最低觀察毒性反應劑量(LOEL)、最小無毒反應劑量(MNTD)以及其他相關參數等信息,可以為評估限量方法的安全性提供科學依據[2]。另外,對藥物不良反應的報告也是評估限量方法安全性的一個重要來源。通過藥品監控數據庫,如世界衛生組織等獲得的大量藥品不良反應報告可以提供藥物使用后出現問題的實例。此外,同類藥物的雜質數據及限量標準也為此類研究提供了參考價值。最后,通過權衡不同雜質的實驗數據、藥物報告以及同類藥物信息等多方面因素,制定該藥品雜質的限量要求和標準,既可確保制藥過程中雜質受到有效控制,提高藥品質量、安全性和療效,又能保證臨床用藥的安全性和有效性。總之,對限量方法的安全性評估應采取多角度、多層次的分析手段,以最大限度地確保藥品的安全性和療效。
3.2長期毒性試驗所允許的最高總雜質暴露
在制定藥品最高允許雜質總暴露標準時,需要綜合考慮多種因素,如雜質毒性試驗、實際應用情況、患者群體等,并兼顧短期和長期的攝入、代謝過程對人體健康的風險影響。例如,在某特定藥物生產過程中,經過長期毒性試驗研究后,最大限度雜質含量需控制在0.15%以內才能確保藥品的安全性和有效性。為了量化雜質暴露,通常需要根據患者在實際治療中可能接受的最大劑量來計算每日雜質暴露量。在制定該指標時,需充分考慮臨床實際用藥的現狀,如給藥途徑(口服、注射、局部等)、劑型(片劑、膠囊、溶液等)以及連續用藥天數等[3]。此外,為了保障不同患者群體的安全,患者的具體情況如年齡、體重、性別、肝腎功能以及合并用藥等因素都應納入綜合評估中,有針對性地設定最高允許雜質總暴露的閾值。借助生物信息學、藥代動力學模型和藥效學分析等先進方法,并通過LC-MS/MS液相色譜-串聯質譜法、ELISA酶聯免疫吸附試驗等技術手段,加強對雜質及藥物代謝產物的識別、測量和評估,對患者暴露的細節進行更精準的掌控。綜上所述,全面考慮長期毒性試驗、實際應用場景以及患者特殊狀況等多個因素對藥品雜質總暴露指標的影響,并綜合運用先進技術和方法進行雜質評估與控制,有利于提高藥品質量和安全性,為患者提供更安全、更有效的藥物治療保障。
3.3雜質用戶限度與生產過程控制限度
雜質用戶限度作為面向終端患者的藥品安全閾值,需要在制定藥品質量標準時全面考慮各階段可能產生的雜質。為確保藥品質量標準的科學性與嚴密性,實現藥品療效、安全性和質量的充分保障,藥品生產過程中需設定相應的過程控制限度,包括在原料藥、化學合成中間體、制藥過程步驟以及輔助物質等環節設置明確的雜質限度標準。在制定這些限度標準時,研究者需要權衡藥品安全性與生產工藝的技術可行性,同時遵循行業內相關法規、藥品質量標準及專業指南,如《中華人民共和國藥典》、ICH指南等。此外,在綜合運用多種檢測方法如高效液相色譜法(HPLC)、氣相色譜法(GC)、核磁共振(NMR)與紅外光譜(IR)等確認和監控雜質生成的過程中,為了確保準確監控藥品生產過程中每個關鍵控制節點的雜質限度,藥廠需要實施嚴格的質量管理體系(如GMP)[4]。確定雜質限度的方式需充分考慮生產現實與藥品質量要求之間的平衡,同時關注生產過程中可能出現的雜質來源以及雜質對不同患者群體的潛在影響。
4結束語
綜上所述,藥品質量標準中雜質限度的確定是一個至關重要的環節,它涉及藥品療效、安全性和質量等諸多方面,為確保藥品的安全使用和高質量,應當充分考慮不同生產階段產生的雜質來源、藥品生產技術可行性以及法規要求等多方面因素。在確定雜質限度時,要嚴格遵循醫藥行業的相關法規、標準及專業指南,并采用先進的檢測技術和方法進行監測和控制。制定實際可行的雜質限度標準,對藥品質量管理、藥物研究開發以及患者健康至關重要。綜合探討藥品質量標準中雜質限度確定方式,不僅有助于提高藥品的療效和安全性,還能為藥品行業的監管和標準制定提供有力支持,幫助實現藥物治療方案的最佳效果,創造更高品質、安全且有效的藥品,為患者提供最優治療方案,最終促進整個醫藥行業的持續發展與進步。
【參考文獻】
[1]田冶,陶曉莎,馮媛媛,等.化學藥品中的遺傳毒性雜質的質量控制[J].中國抗生素雜志,2024,49(1):13-25.
[2]田向斌,柯靜,王文麗,等.鹽酸羥考酮原料藥中元素雜質控制方法研究[J].中國藥業,2023,32(21):108-112.
[3]張蕓,王亞敏.基于ICH Q3D(R2)解讀藥品元素雜質研究的基本考慮[J].中國新藥雜志,2023,32(17):1719-1724.
[4]王靖,張志新.藥品元素雜質控制政策解讀:超出PDE的可接受限度的建立[J].臨床醫藥文獻電子雜志,2019,6(56):196-197.
【作者簡介】
闞洪峰,女,1987年出生,工程師,碩士,研究方向為藥品質量研究。
通信作者:宋錫瑞,男,1988年出生,工程師,碩士,研究方向為創新藥物研發,1043058756@qq.com。
(編輯:劉一童)
