利用CRISPR/Cas9基因編輯片段敲除技術創制玉米多樣化等位變異
2024-10-16 00:00:00張茂林王娟嚴佳麗何春梅徐倩劉鐵山董瑞劉春曉關海英劉強汪黎明曾廷儒
山東農業科學 2024年9期

摘要:利用CRISPR/Cas9基因編輯技術創制等位變異已經成為眾多植物中增加種質資源多樣性的重要手段。提高玉米等谷物中類胡蘿卜素含量是發展中國家解決維生素A缺乏癥的一種經濟方法。本研究以玉米中調控類胡蘿卜素合成的基因crtRB1為研究對象,利用雙靶標片段缺失性基因編輯技術,在原生質體中對crtRBI的啟動子順式元件進行片段敲除,獲得了具有片段缺失和InDel(插入或缺失)的多樣化等位變異,這為在玉米中實現穩定的crtRB1啟動子元件的缺失突變奠定了基礎,對啟動子元件的功能研究和玉米的品質改良具有重要意義。
關鍵詞:玉米;crtRBI;基因編輯;片段敲除;啟動子
中圖分類號:Q781:S503.531+3 文獻標識號:A 文章編號:1001-4942(2024)09-0001-05
糧食安全是世界首要問題,隨著人口不斷增長,現有的糧食產量和品質越來越不能滿足人們的需求。玉米是三大作物之一,是我國乃至世界種植面積最大、產量最高的作物。玉米不僅是糧飼兼用作物,也是能源產品的重要原料植物。玉米傳統育種近幾十年取得長足的進步,但主要基于現存的自然變異進行人工選擇,再通過雜交聚合選育新品種,費時費力。然而,現存的變異遠遠不能滿足現代育種的需求,這就需要用生物技術手段人工創制遺傳變異新種質,以縮短育種周期,提高育種效率。
近年來,基因編輯技術的研究和發展突飛猛進,新興的CRISPR/Cas9基因編輯技術為育種家提供了能夠精確創造等位基因的大好機遇。CRISPR/Cas9基因組編輯技術最早于2010年在原核免疫系統中被發現,也是繼第一代鋅指結構核酸酶(ZFNs)和第二代轉錄激活子樣效應因子核酸酶技術(TALENs)之后,被公認的第三代基因組編輯技術。植物主要通過非同源末端連接(NHEJ)途徑來修復雙鏈DNA斷裂缺口(DSBs),以此進行堿基的敲除和敲入,此過程可以導致基因移碼突變,使目的基因功能缺失,從而達到敲除基因的目的。
CRISPR/Cas9基因組編輯技術在2013年第一次應用于植物中,之后便成為植物科學界的明星技術被廣泛應用,如擬南芥、玉米、水稻、大豆、番茄等。至此,CRISPR/Cas9在農作物中的應用已非常流行,近幾年用在農作物上創造了很多有益變種,涉及產量、品質、抗病和抗逆等。CRISPR/Cas9技術操作簡單,編輯效率高,可用來創造多倍體植物的突變體、基因組大片段的敲除以及產生多基因突變體等。當同一條染色體相距一定距離的兩個靶位點同時被編輯時,靶標之間的片段有一定的幾率被敲除。在模式植物擬南芥中,Zhao等通過此方法將含有多個基因的基因組大片段成功敲除:另外,水稻中就有長達245 kb的基因組片段用此方法被剪切掉。Rodriguez-Leal等利用CRISPR/Cas9基因組編輯技術對SICLV3啟動子順式元件進行片段敲除,創造了多樣化的等位變異,并篩選得到了具有連續表型的多樣化順式元件突變體,并可以直接作為育種的種質資源,為分子育種提供了新的思路和方法,使得通過改造玉米啟動子獲得理想的變異新種質成為可能。這些等位突變如果用傳統的方法需要花費大量的時間和精力去篩選,而這種基于CRISPR/Cas9基因組編輯技術的啟動子變異方法成功繞開了傳統育種的限制,直接創造并且篩選獲得了科學家渴望得到的變異,為突破分子育種瓶頸提供了新的思路。
crtRBI是調控玉米籽粒中維生素A含量的關鍵基因,該基因中一個稀有變異降低了基因的轉錄水平,大幅提高了玉米籽粒中維生素A的含量。此舉惠及患有維生素A缺乏癥的數萬非洲兒童。本研究利用CRISPR/Cas9基因編輯技術對玉米crtRB1基因的啟動子區域進行編輯,旨在進一步明確crtRBI的功能,同時為有效調節玉米籽粒中維生素A的含量提供理論參考。
1材料與方法
1.1試驗材料
本研究所用植物CRISPR/Cas9基因編輯載體pCPB-ZmUbi-hspCas9由謝傳曉研究員提供。DL2000 DNA Marker、T4連接酶、DNALoading Buffer、DNA Marker、FastPfu、2x Taq Mix、大腸桿菌DH5a、EHA105農桿菌感受態細胞等購自北京全式金生物技術有限公司:限制性內切酶BsaI購自紐英倫生物技術(北京)有限公司;高保真酶購于南京諾唯贊生物科技股份有限公司:氯仿,無水乙醇、異丙醇等試劑均購于國藥集團。引物由青島擎科梓熙生物技術有限公司合成,基因測序由上海生工生物工程技術服務有限公司完成。
1.2雙靶位點設計
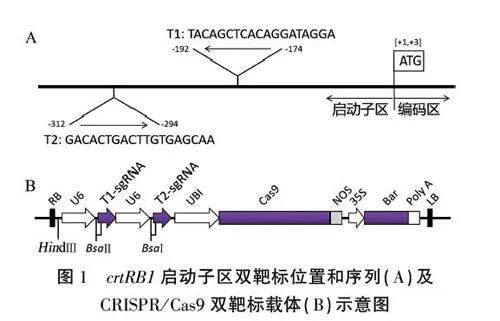
由于轉化所用受體自交系KN5585的基因組和B73的基因組有一定的差異,我們首先以B73基因組做參考,對選取的需要進行片段敲除的序列兩端設計引物PRB/-T-F和pRBl-T-R(表1),然后以KN5585的DNA為模板擴增測序確定要進行片段敲除的啟動子區域序列。通過CRISPR-P 2.0在線設計網站(http://crispr.hzau.edu.cn/cgi-bin/CRISPR2/CRISPR)設計crtRBI啟動子目標區域的2個靶位點Tl和T2(圖1A)。
1.3片段敲除載體構建
1.3.1靶標引物退火形成雙鏈片段 靶標T1對應的兩個引物是pRBI-T1-F和PRB/-T1-R,靶標T2對應的兩個引物是pRB/-T2-F和pRB1-T2-R,將每個靶標對應的兩條引物分別成對混合,直接退火形成雙鏈:37℃20 min,95℃5min,以6℃/min的速度降至25℃,形成帶粘性末端Bsa I酶切位點的靶標片段,備用。
1.3.2sgfRNA-U6片段的獲得 以pCPB-ZmUbi-hspCas9質粒為模板,用引物sgU6Fl/sgRNA-R擴增片段sgRNA,用U6-F/sgU6R1(表1)擴增U6片段,然后將兩個PCR產物混合后為模板,利用兩個片段之間的重疊序列,以引物sgU6F1/sgU6R1(表1)(帶BsaI酶切位點)進行重疊PCR。反應體系:上、下游引物(10 umol/L)各2uL,2x Buffer 25 uL,dNTP Mix(10 mmol/L)1uL,兩個模板產物均為1 uL,PhantaMax高保真DNA聚合酶1uL,用ddH2O補至50 uL。反應程序:95℃1min;95℃15 s、55℃15 s,72℃30 s,35個循環;72℃5min。電泳,選擇合適大小的條帶切膠回收,回收產物用BsaI酶切純化,獲得帶BsaI粘性末端的sgRNA-U6片段,備用。
1.3.3載體線性化 載體pCPB-ZmUbi-hspCas9中具有毒性致死基因ccdb,使用限制性內切酶BsaI將載體中的ccdb切除,形成兩端具有BsaI粘性末端的線性化載體,備用。
1.3.4連接形成最終載體 構建敲除載體的每個靶位點分別由獨立的U6啟動子驅動(圖1B)。將上述獲得的T1和T2靶標片段、sgRNA-U6片段、線性化載體pCPB-ZmUbi-hspCas9用T4連接酶進行鏈接組裝,體系為:3個片段各0.5 uL,T4連接酶0.3 uL,加水至5uL。25℃連接1h。將連接產物轉化大腸桿菌DH5a,之后于LB平板培養基(含Kan)上培養,挑取單克隆用引物BGK-T-F/BGK-T-R(表1)進行菌落PCR鑒定及測序,比對篩選正確的克隆提取質粒。
1.4玉米原生質體的提取和轉化
參考文獻中玉米原生質體的制備及轉化方法并稍作調整。將玉米種子在花盆中培養(25℃,16L:8D)8 d左右,取幼苗葉中間段切成寬0.5-1.0 mm的細條,轉移到酶解液[含0.6mol/L甘露醇、10 mmol/L MES(pH值5.7)、1.5%纖維素酶R10、0.75%離析酶、10 mmol/L CaCl2.0.1% BSA];用錫箔紙包住后放置28℃搖床,50r/rmn避光酶解5 h:加入等體積W5(含154mmol/L NaCI、125 mmol/L CaCL2、5 mmol/L KCl、2mmol/L MES),輕柔搖晃均勻后過40 um細胞篩:加入MMG溶液(含0.4 mol/L甘露醇、15mmol/L MgCl2、4 mmol/L MES)重懸原生質體;將構建好的質粒利用PEG介導的方法轉入原生質體,避光條件下28℃培養48 h:離心收集玉米原生質體,用DNA快速提取試劑盒提取基因組DNA。
1.5基因編輯方式檢測
以提取的原生質體DNA為模板,使用特異性引物pRB1-T-F/pRBl-T-R進行PCR擴增,將PCR產物純化后連接pEASY-Blunt載體,并將連接產物轉化大腸桿菌DH5a,涂板LB固體培養基(Kana),37℃倒置培養過夜。等菌落長到合適大小后,選取大于50個獨立的菌落分別進行PCR擴增,將擴增產物送至上海生工生物工程技術服務有限公司測序。通過測序結果比對判定兩個靶位點之間有無大片段缺失以及兩個靶位點各自區域的編輯情況(插入、替換或缺失)。
2結果與分析
2.1片段敲除載體的構建及鑒定
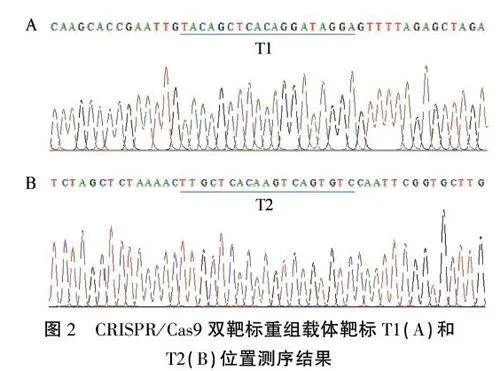
以構建好的片段敲除載體質粒為模板,PCR擴增后分別用上游和下游引物測序,通過比對測序結果中靶標T1(BGK-T-F測序)和T2(BGK-T-R測序:序列反向互補)以及側翼序列,篩選獲得了預期的重組載體(圖1B,圖2),證明所需的片段敲除載體構建成功。
2.2原生質體轉化及編輯情況檢測
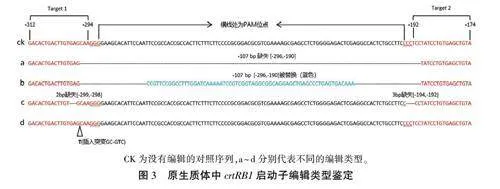
對原生質體DNA進行PCR擴增,產物連接pEASY-Blunt載體后轉化大腸桿菌DH5a,長出菌點后,選取獨立的50個進行菌落PCR擴增并測序。通過對測序結果進行比對(圖3)發現,目標編輯區域發生了大片段缺失和替換,同時,兩個靶標各自位點也發生InDel(替換、插入或缺失)。發生編輯的位點大部分是雙等位基因,只有6%(50個中有3個)包含一個野生型拷貝。雙靶標之間發生大片段缺失的概率為8%(圖3a),發生大片段替換的概率為2%(50個中有1個)(圖3b)。兩個靶標各自位點也有InDel,T1位點發生的編輯類型包括2 bp缺失(圖3e)和單堿基(T)插入(圖3d),概率分別為48%和16%;T2位點發生了3 bp的缺失(圖3e),概率為32%。對于單個突變體來講,兩個靶標位點編輯方式都有InDel的概率為8%(圖3e),只有一個靶標位點區域有InDel的概率為8%(圖3d)。表明本研究所用的CRISPR/Cas9基因編輯系統編輯效率較高,能夠滿足片段敲除和多樣化等位變異的需求。
3討論與結論
在育種過程中,經常遇到需要微調某種性狀來尋找眾多育種性狀的最優平衡點,從而達到品種選育與改良的目的。基因編輯技術的突飛猛進給育種帶來了重大機遇。利用CRSIPR/Cas9雙靶標基因編輯技術,對啟動子的順式作用元件進行片段缺失性突變,來調節基因的功能,能夠在創制新的等位突變的同時,微調相應性狀,從中選擇最優的新種質資源應用到育種中,從而達到品種改良的目的。
本研究將構建的CRSIPR/Cas9雙靶標片段敲除載體轉入玉米原生質體,通過編輯方式和效率檢測,證明該系統的有效性,能夠滿足片段敲除的需求,同時編輯方式的多樣性具備創制多樣化變異材料的條件。本研究結果可為評估啟動子順式元件的功能及其在育種中的價值提供理論參考,也為發展一套全面詳細研究啟動子順式元件的方法以及評估啟動子不同調控元件在育種中的應用潛力提供理論支撐。至此,通過人工改造順式元件來調控基因功能成為可能,作物遺傳改良已經進入一個新的時代,需要我們共同探索。
