包封骨形態發生蛋白2 的不同相對分子質量聚乳酸-聚乙醇酸共聚物微囊促進成骨效果的研究
2024-10-19 00:00:00袁莉紅陳晨馬語笛梁睿貞
華西口腔醫學雜志 2024年5期


[摘要] 目的 探索不同相對分子質量聚乳酸-聚乙醇酸共聚物(PLGA) 微囊包封骨形態發生蛋白2 (BMP-2)對成骨細胞骨形成能力的影響。方法 用雙通道微量注射泵制備包封BMP-2 的2 種相對分子質量(12 000 和30 000) 的PLGA微囊。用光學顯微鏡及掃描電子顯微鏡(SEM) 觀察微囊形態結構;磷酸鹽緩沖溶液浸泡法表征微囊緩釋性能;細胞Calcein-AM/PI 染色及CCK-8 法檢測微囊細胞相容性;Transwell 遷移實驗檢測包封BMP-2 的微囊作用48 h 對MC3T3-E1 細胞的趨化作用;堿性磷酸酶活力測定、茜素紅染色法檢測微囊作用MC3T3-E1 細胞后對細胞骨形成能力的影響。結果 2 種相對分子質量的微囊均表面光滑,具有良好的細胞相容性。相對分子質量12 000 的微囊的趨化作用最佳。相對分子質量30 000 的微囊較相對分子質量12 000 的微囊緩釋時間長,且初始爆發量減少了約25%。成骨誘導14、21 d 后,相對分子質量30 000 的微囊形成的鈣沉積結節較相對分子質量12 000 的微囊多。結論 本研究通過調控PLGA的相對分子質量控制BMP-2 的釋放,發現相對分子質量30 000 的微囊能夠更好地誘導MC3T3-E1 細胞的長期骨形成能力。
[關鍵詞] 聚乳酸-聚乙醇酸共聚物微囊; 骨形態發生蛋白2; 成骨
[中圖分類號] R318.08 [文獻標志碼] A [doi] 10.7518/hxkq.2024075
組織工程支架能夠支持和促進細胞生長,是當前組織缺損修復最重要的干預方法[1]。骨形態發生蛋白2 (bone morphogenetic protein 2,BMP-2)是一種在骨和軟骨發育、修復中發揮關鍵作用的轉化生長因子,常被添加于骨組織工程支架中[2],已被美國食品藥品監督管理局批準在骨科和牙科中臨床應用[2-3]。但是,BMP-2 的大劑量應用不僅價格昂貴,還會導致異位骨化、炎癥反應、軟組織水腫、破骨細胞活化等嚴重并發癥的發生。同時,BMP-2 在體內會快速降解,半衰期短[4-5]。研究[3]表明,BMP-2 至少需在數周的時間段內保持最小劑量水平才能有效[6-7]。針對此情況,目前最常見解決方式是通過能夠緩釋藥物的支架來局部遞送BMP-2。
支架是用來支持細胞生長的結構,常見形式包括多孔的3D 基質、水凝膠、球體和纖維網[1]。支架材料主要有金屬、陶瓷、聚合物等。其中聚乳酸-羥基乙酸共聚物[poly(lactic-co-glycolic acid),PLGA] 這種合成聚合物具有良好的生物相容性,已獲得美國食品藥品監督管理局的生物醫學應用批準。PLGA是一種可生物降解的脂肪族無定形聚合物,由乳酸(lactic acid,LA) 和乙醇酸(glycollic acid,GA) 兩種單體聚合而成,其LA∶GA配比、相對分子質量、分子鏈端基類型等都可以影響其降解速率[8-9]。Wei 等[2]通過將BMP-2 負載于介孔羥磷灰石微球和PLGA組成的復合支架中使BMP-2 緩釋,該支架與骨髓間充質干細胞(bonemarrow mesenchymal stem cells,BMSCs) 共培養,實時定量逆轉錄聚合酶鏈反應表明BMP-2 緩慢、長期釋放有利于成骨相關標志物的長期高表達。
目前通過支架遞送BMP-2 的方式主要包括與支架共價結合、在支架中被非共價固定、包封在支架中等。其中,將BMP-2 包封于顆粒中這種方式,由于可以將BMP-2 在釋放前與體內環境隔離,已經成為研究的一個熱點[4]。但是,目前用于緩釋生長因子的支架存在突釋嚴重、釋放時間短的缺點[3]。因此,制備一種能夠減少突釋,在未釋放前將生長因子與體內環境隔離,并能夠盡可能長時間維持其生物活性的支架,是現在研究的熱點。本研究基于BMP-2 緩釋的目的,使用PLGA 微囊包封BMP-2,并對2 種不同相對分子質量(12 000和30 000) 微囊中BMP-2 的釋放速率及其對成骨細胞骨形成能力的影響進行探討。
1 材料和方法
1.1 PLGA微囊的制備
取8 g 聚乙烯醇(polyvinyl alcohol,PVA) 溶于400 mL 去離子水,制備成2%的PVA 溶液。將PVA 溶液與磁力攪拌子一起盛于500 mL 燒杯中,置于磁力攪拌器[MS-H-Pro+,大龍興創實驗儀器(北京) 有限公司] 上備用。取1 g PLGA溶于10 mL二氯甲烷,配置成10%的PLGA溶液,置于50 mL 注射器中。將需要包封的內容物溶液裝入20 mL注射器,包封的內容物有去離子水、牛血清白蛋白(bovine serum albumin,BSA) 溶液(Bio‐Froxx 公司, 德國)、BMP-2 溶液(500 ng/mL)(蘇州近岸蛋白質科技股份有限公司) [10-11],根據實驗需要選擇。將裝有PLGA溶液的注射器與裝有需要包封的內容物溶液的注射器分別安裝到雙通道微量注射泵(SP-2000,寧波安諾醫療器械科技有限公司) 的A、B 通道,調整A、B 通道注射速度分別為580、80 mL/h,連接同軸針頭的外針和內針入口,將同軸針頭置于PVA 溶液液面以下,調整磁力攪拌器轉速,190、270 r/min 分別對應相對分子質量為12 000、30 000 的PLGA溶液。注射器排氣后開始注射。注射結束后,將針頭從PVA溶液中取出。攪拌8~12 h 后,靜置燒杯,使PLGA微囊沉淀,隨后棄去大部分上清液,加入去離子水清洗3 遍,冷凍干燥12 h。
1.2 PLGA微囊的形態觀察及粒度分析
通過掃描電子顯微鏡(scanning electron microscope,SEM) 在10 kV電壓下觀察微囊的形態。通過光學顯微鏡觀察微囊形態,拍照記錄后用ImageJ測量微囊的粒徑(n=300)。
1.3 PLGA微囊的包封率檢測
制備PLGA微囊時,在去上清液前,從燒杯中取出2 mL PVA溶液,加入0.5 g 固體無水硫酸鈉,渦旋10 min,然后以4 000 r/min 的轉速離心10 min。上清液中含有未被包裹在微囊內的BSA,用超微紫外分光光度計(NanoDropOne,ThermoFisher‐Scientific 公司,美國) 檢測上清液中的BSA 濃度(Cx)。假設用于包封去離子水微囊的PVA溶液的BSA 濃度為基礎濃度(C0),通過Cx 減去C0 來計算PVA 溶液中未被包裹的BSA 的實際濃度。使用下列公式計算包封率:Ma (mg) = (Cx?C0)×400 (mL);包封率(%) =Mi-Ma/Mi×100% 。其中,Mi 為制備內容物為BSA溶液的微囊時總共使用的BSA的量,Ma 為實際流失在PVA溶液中的BSA的總量。
1.4 磷酸鹽緩沖溶液(phosphate buffer solution,PBS) 浸泡法表征微囊緩釋性能
稱取2 種相對分子質量(12 000 和30 000)、包封內容物分別為BSA 和去離子水的PLGA 微囊50 mg,浸入100 μL PBS (pH=7.4) 中,并在37 ℃恒溫振蕩器(ZQZY-78AV,上海知楚儀器有限公司) 中以200 r/min 振蕩。用超微量紫外分光光度計測定包封BSA微囊的PBS 中的BSA濃度,并將該量設為Cx,每5 d 更換一次新鮮的PBS。假設包封去離子水微囊的PBS 中的BSA濃度為基礎濃度(C0)。使用下列公式計算實際釋放的BSA 量(Mt):Mt (μg) = (Cx?C0)×100 (μL);使用下列公式計算BSA 的實際釋放率(release rate,RR):RR (%) =Mt/Me×100% 。其中,Me 為50 mg 微囊中BSA的總量。
1.5 細胞Calcein-AM/PI 染色及CCK-8 法檢測微囊細胞相容性
分別將2 種相對分子質量(12 000 和30 000)包封BMP-2 的PLGA 微囊40 μL 加入24 孔板中,紫外照射2 h 進行滅菌,PBS 清洗一遍,然后將微囊浸泡在1 mL 完全培養基中24 h 進行預培養。以不加任何刺激的孔為空白對照組。將MC3T3-E1 細胞按5×104 個/孔的密度接種到孔板中,于37 ℃、5%CO2和100%濕度的細胞培養箱中進行培養,每2 d 更換一次培養液,5 d 后用Calcein-AM/PI 細胞活性與細胞毒性檢測試劑盒(上海碧云天生物技術股份有限公司) 按照說明書對細胞進行染色,倒置熒光顯微鏡(Leica 公司,德國) 下觀察染色結果。
分別將2 種相對分子質量(12 000 和30 000)包封去離子水或BMP-2 的PLGA微囊20 μL加入96孔板中,紫外照射2 h 進行滅菌,PBS 清洗一遍,然后將微囊浸泡在完全培養基(含10%FBS 和1%青霉素/鏈霉素) 中24 h 進行預培養。以不加任何刺激的孔為空白對照組。將MC3T3-E1 細胞接種到孔板中(接種密度5 000 個細胞/孔),于37 ℃、5%CO2和100%濕度的細胞培養箱中進行培養,每2 d 更換一次培養基。在第1、3、5、7 天分別吸除舊培養基,向每孔中加入150 μL 含10%CCK-8 溶液(APExBIO 公司,美國) 的基礎培養基,避光孵育2 h 后吸取100 μL 液體置于另一96 孔板,使用酶標儀(SpectraMax M2e,Molecular Devices 公司,美國) 于450 nm 波長下檢測光密度(opticaldensity,OD)。
1.6 Transwell 遷移實驗檢測細胞趨化
制備緩釋微囊: 分別將2 種相對分子質量(12 000 和30 000) 包封BMP-2 的PLGA 微囊加入24 孔板中,加入1 mL PBS,37 ℃恒溫振蕩器以200 r/min 的轉速振蕩3 d 后吸除上清液,PBS 清洗一遍,凍干,設為BMP-2 scaffolds (sustained release)組,即BMP-2 scaffolds SR組。
取20 μL 緩釋微囊加入24 孔板的Transwell 小室的下室,另取2 種相同量未緩釋微囊(即按照1.1 制備的包封BMP-2 的PLGA 微囊) 加入另外的孔中進行對照,設為BMP-2 scaffolds (non-sustained-release) 組,即BMP-2 scaffolds 組。
所有樣品紫外照射2 h 滅菌后,PBS 清洗一遍,并加入1 mL基礎培養基預培養24 h。第2天吸除舊培養基,PBS清洗一遍,在下室中加入700 μL基礎培養基,上室中加入200 μL 基礎培養基重懸后的MC3T3-E1 細胞,接種密度為2×105個/室。每組設置3 個復孔。培養48 h 后使用4% 多聚甲醛(Biosharp,北京蘭杰柯科技有限公司) 固定細胞,并用棉球小心擦去上室細胞,使用結晶紫染色液(上海碧云天生物技術股份有限公司) 對下室細胞進行染色,并在倒置熒光顯微鏡下觀察細胞遷移結果。隨后對下室細胞進行半定量分析,在小室中加入700 μL 33%冰乙酸,37 ℃靜置20 min,使下室結晶紫充分溶解,在595 nm 波長處檢測OD值。
1.7 微囊的堿性磷酸酶(alkaline phosphatase,AKP)活力測定
按照1.6 中的方法制備緩釋微囊,分別取2種相對分子質量(12 000 和30 000) 的緩釋微囊40 μL 加入6 孔板中,設為BMP-2 scaffolds SR 組。取2 種相同量未緩釋微囊(即按照1.1 制備的包封BMP-2 的PLGA 微囊) 加入另外的孔中, 設為BMP-2 scaffolds 組,并設置一個不加任何刺激的孔作為空白對照組。所有樣品紫外照射2 h 滅菌后,PBS 清洗一遍,加入2 mL 完全培養基預培養24 h。
將MC3T3-E1 細胞以3×105個/孔的密度接種于6 孔板。當細胞密度為70%時,用成骨誘導培養基誘導細胞成骨分化,培養基的配制成分為α-MEM+10%FBS+1%青鏈霉素+10 mmol/L β-甘油磷酸鈉+0.2 mmol/L 抗壞血酸+100 nmol/L 地塞米松。每2~3 d 更換1 次培養基,培養7 d 后,冰上吸棄各組舊培養基,PBS 洗2 遍。用RIPA 裂解液(上海碧云天生物技術股份有限公司) 提取總蛋白,BCA蛋白測定試劑盒(上海碧云天生物技術股份有限公司) 定量,按照AKP 試劑盒(南京建成生物工程研究所) 說明書測定各組樣本的AKP 活力。
1.8 浸提液誘導成骨分化
制備緩釋微囊的浸提液:分別將2 種相對分子質量(12 000 和30 000) 包封BMP-2 的PLGA微囊200 mg 放入2 mL 凍存管中,紫外照射2 h 滅菌,PBS 清洗1 遍。加入1 mL 完全培養基,密封后置于37 ℃恒溫振蕩器上以200 r/min 振蕩3 d,吸去舊培養基,PBS 清洗1 遍,加入1 mL 新鮮的完全培養基,置于37 ℃恒溫振蕩器上以200 r/min 振蕩。此后每72 h 靜置凍存管內液體并提取上清液,以1∶20 的比例加入成骨誘導培養基中。之后加入1 mL 新鮮的完全培養基繼續振蕩。將該種培養液培養的細胞設為BMP-2 scaffolds (sustained release)組,即BMP-2 scaffolds SR組。
制備未緩釋微囊的浸提液:稱取2 種相對分子質量(12 000和30 000) 包封BMP-2 的微囊200 mg于凍存管中,紫外照射2 h 滅菌,PBS 清洗一遍,加入1 mL完全培養基,置于37 ℃恒溫振蕩器上以200 r/min 振蕩。此后每72 h 靜置凍存管內液體并提取上清液,以1∶20 的比例加入成骨誘導培養基中。之后加入1 mL 新鮮的完全培養基繼續振蕩。
將該種培養液培養的細胞設為BMP-2 scaffolds(non-sustained-release) 組,即BMP-2 scaffolds 組。將MC3T3-E1 細胞以3×105個/孔的密度接種于6 孔板。當細胞生長至70%匯合狀態時,對照組用成骨誘導培養基,實驗組用上述方法制成的浸提液培養基誘導成骨分化,成骨誘導培養基成分為α-MEM+10%FBS+1%青鏈霉素+10 mmol/L β-甘油磷酸鈉+0.2 mmol/L 抗壞血酸+100 nmol/L 地塞米松。每3 d 更換一次培養基。
1.9 微囊浸提液的AKP 活力測定
按照1.8 中的方法培養MC3T3-E1 細胞,培養7 d 后,冰上吸棄各組舊培養基,PBS 洗2 遍。用RIPA裂解液提取總蛋白,BCA蛋白測定試劑盒定量,按照AKP 試劑盒說明書測定各組樣本的AKP活力。
1.10 茜素紅染色
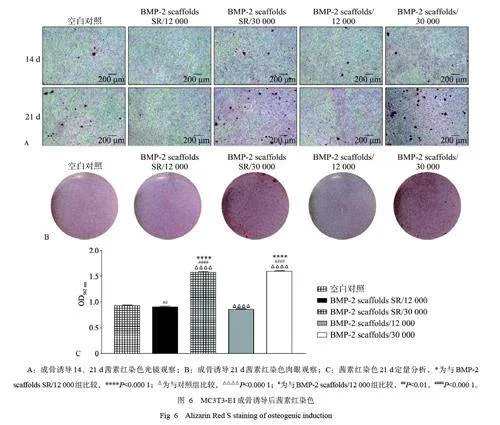
按照1.8 中的方法培養MC3T3-E1 細胞,成骨誘導14、21 d 后,用1%茜素紅S 染色液(pH4.2)(北京雷根生物技術有限公司) 進行礦化檢測。檢測完畢后使用2%氯化十六烷基吡啶溶液(上海阿拉丁生化科技股份有限公司) 孵育30 min,使用酶標儀在562 nm波長處檢測OD值。
1.11 統計學方法
采用SPSS 26 軟件進行統計學分析。用均數±標準差表示數據,多組間比較采用單因素方差分析,兩組間比較采用獨立樣本t 檢驗,Plt;0.05 認為差異有統計學意義。
2 結果
2.1 PLGA微囊的形態及粒度
制備的PLGA微囊形態及粒度表征見圖1,結果顯示:PLGA微囊表面光滑,大多數粒徑集中在80~160 μm,相對分子質量30 000 的PLGA微囊粒徑較相對分子質量12 000 的微囊粒徑略大。
2.2 微囊的包封率
相對分子質量為12 000 和30 000 的PLGA微囊的包封率分別為24.52%±1.53%、46.32%±6.07%。統計分析表明,相對分子質量為30 000 的微囊包封率高于相對分子質量為12 000 的微囊,差異有統計學意義(P=0.004)。
2.3 微囊的緩釋性能
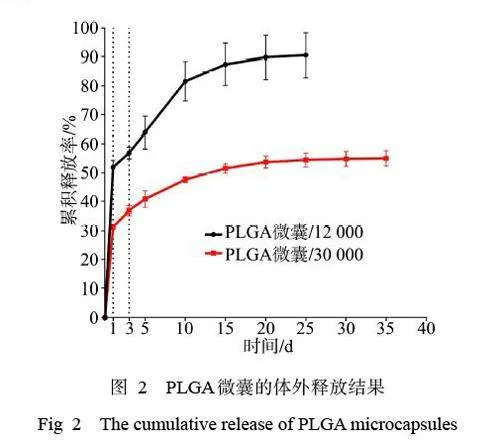
2 種相對分子質量PLGA制備的包裹BSA溶液的微囊釋放性能見圖2,結果顯示:這2 種微囊均可以持續釋放BSA,且第1 天都表現出了初始爆發性釋放。其中,相對分子質量12 000 的微囊第1 天釋放率接近55%,10 d后釋放速率顯著減緩,第25天時幾乎檢測不到BSA的釋放,累積釋放率已達到90%;相對分子質量30 000 的微囊第1 天釋放率約為30%,其后緩慢持續釋放,第35 天仍能檢測到BSA 的釋放,累積釋放率約為50%。與相對分子質量12 000 的微囊相比,相對分子質量30 000的微囊的緩釋時間更長,且初始爆發量減少了約25%。
2.4 細胞相容性
Calcein-AM/PI 染色(圖3A) 顯示,加入PLGA微囊的實驗組與空白對照組細胞均能正常增殖,并且實驗組細胞可以黏附到PLGA微囊表面,使PLGA微囊的輪廓變亮。相對分子質量12 000 的微囊降解速率較快,降解后黏附于孔板底部,相對分子質量30 000 的微囊在第5 天時僅有少部分黏附于底部。CCK-8 結果(圖3B、C) 顯示了空白對照組以及加入PLGA微囊的實驗組1、3、5、7 d的細胞活力,可見實驗組成骨細胞有隨時間增殖的趨勢,2 種不同相對分子質量的PLGA微囊對成骨細胞均具有良好的生物相容性。
2.5 細胞趨化
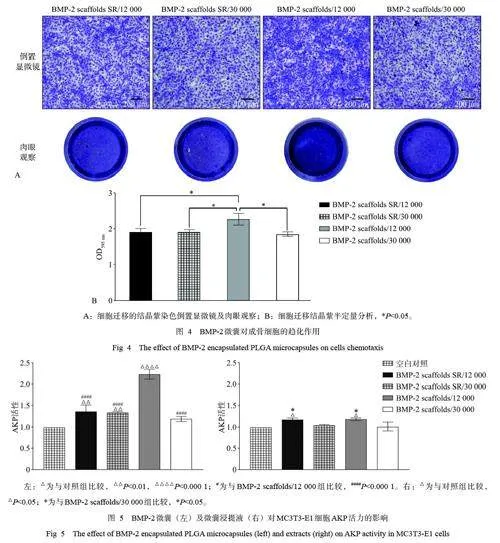
各組下室細胞染色以及半定量結果(圖4) 顯示: BMP-2 scaffolds/12 000 組的趨化作用最佳(Plt;0.05),能吸引更多細胞向其遷移。
2.6 微囊的AKP 活力
在成骨誘導培養基中培養7 d 后,AKP 活性測定結果(圖5 左) 顯示,BMP-2 scaffolds/12 000 組相對AKP 活力最高,表明BMP-2 從微囊中釋放后仍能保持活性,發揮成骨分化作用,同時從側面表明BMP-2 scaffolds/12 000 組在7 d 內釋放的藥物較另外3 組更多,這與2.3 中得到的緩釋結果相一致。2 個BMP-2 scaffolds SR 組AKP 活力差異無統計學意義,但都高于空白對照組。表明2 種微囊經過3 d 的緩釋仍能釋放出BMP-2, 并能夠保持BMP-2 的活性。BMP-2 scaffolds/30 000 組微囊的AKP 活力與空白對照組差異無統計學意義。
2.7 浸提液對MC3T3-E1 體外成骨分化的影響
微囊浸提液的AKP 活性測定結果(圖5 右)顯示,相對分子質量12 000 的微囊的AKP 活力高于相對分子質量30 000 的微囊(Plt;0.05),而相對分子質量30 000 的微囊的AKP 活力與空白對照組差異無統計學意義(Pgt;0.05)。
將MC3T3-E1 細胞在成骨誘導培養基中培養,茜素紅染色及半定量分析結果見圖6。相對分子質量30 000 的2 組較其他組鈣沉積結節多,相對分子質量12 000 的2 組鈣沉積結節較空白對照組少,BMP-2 scaffolds SR/12 000 組幾乎無鈣沉積結節。14、21 d 時茜素紅染色結果對比,空白對照組以及相對分子質量30 000 的2 組鈣沉積結節都隨時間增多,但是相對分子質量12 000 的2 組鈣沉積結節21 d 時與14 d 時差異無統計學意義。21 d 時茜素紅半定量分析顯示,相對分子質量30 000 的2 組鈣沉積結節高于空白對照組以及相對分子質量12 000的2 組(Plt;0.05)。
3 討論
本課題組的前期研究利用相對分子質量為12 000 以及150 000 的PLGA 微囊制備了一種可以持續釋放生長因子的支架。研究[10]表明,相對分子質量為12 000 的微囊在14 d 內促進骨形成的能力優于相對分子質量為150 000 的微囊。然而,骨形成在體內是一個長期的過程,BMP-2 需要在體內釋放至少數周。本研究基于上述實驗結果,選擇制備相對分子質量為12 000 以及30 000 的微囊進行對比,在其內包裹BSA研究釋藥能力。結果表明,相對分子質量為12 000 的微囊釋放BSA 的時間持續約25 d, 其中第1 天約釋放其含量的55%;相對分子質量為30 000 的微囊在緩釋35 d時僅釋放了其所載藥物的50%,第1 天釋放了其含量的30%。BMP-2 通過初始爆發釋放,隨后緩慢持續釋放的方式遞送能更好地促進骨形成[12]。同時釋放動力學結果表明,相對分子質量為12 000的微囊在初期釋放的BMP-2 含量最高,BMP-2 的作用為促進成骨細胞的增殖和分化[13-14],在細胞趨化實驗中BMP-2 也表現出了對成骨細胞的趨化作用,這印證了前期課題組的研究結果[10]。
本研究發現,微囊的粒徑大多數集中在80~160 μm。支架的孔隙gt;50 μm有利于細胞的進入以及血管生成[15]。Rmaidi 等[16]測定了PLGA對細胞的黏附,光學顯微鏡顯示將細胞與材料共培養4 h 后細胞能夠黏附并擴散在材料表面,并且黏附的細胞總數約為接種總數的90%。本研究微囊的Calce‐in-AM/PI 染色結果表明,5 d 時有大量的MC3T3-E1 黏附于微囊的周圍,在微囊的邊緣形成一個粗光圈,這是由于PLGA的疏水性可以使細胞迅速黏附至其表面。在這種微囊內部包裹BMP-2,通過AKP 活力檢測、茜素紅染色及半定量分析證明了BMP-2 被包裹后通過微囊緩釋能夠起到促進成骨的效果,同時從側面證明BMP-2 通過微囊釋放后仍能保持其活性。但是相對分子質量為12 000 的微囊雖然在7 d 的AKP 活性檢測實驗中表現出明顯的誘導成骨能力,但是在14、21 d 的長期誘導成骨實驗中表現出了明顯的不足,甚至成骨能力低于空白對照組。出現這種現象,一方面是由于相對分子質量為12 000 的微囊在前期突釋嚴重,BMP-2 已早期大量釋放;另一方面,PLGA是通過酯鍵水解降解為乳酸和羥基乙酸,其酸性降解產物會引發PLGA自催化作用,加速其降解,并使局部微環境呈弱酸性。PLGA的水解過程分為兩個階段,第一階段PLGA的相對分子質量逐漸降低,而總重量不變;第二階段PLGA相對分子質量降低,水解產物溶于水,總重量快速下降[8-9,17]。由于相對分子質量為12 000 的微囊的相對分子質量較小,導致在成骨初期就已到達水解的第二階段。過快的水解導致周圍環境呈弱酸性,周圍組織出現炎癥反應,阻礙了成骨分化的進程[8,18]。
本研究初步證明,通過調控PLGA相對分子質量的大小,可以控制其內包封藥物的釋放速率。與相對分子質量為12 000 的PLGA微囊相比,相對分子質量為30 000 的PLGA微囊對誘導長期的成骨分化更有利。但是本研究僅局限于體外研究,并且由于本研究目的在于比較2 種相對分子質量微囊的釋藥能力對成骨效果的影響,應用了微囊的浸提液,對于該微囊的形態結構對MC3T3-E1 成骨分化的影響體現不足,因此下一步需要改進實驗方法,并增加體內實驗,進一步驗證該結論的準確性。同時,在實際實驗中發現低相對分子質量的微囊在緩釋的后期會使周圍環境呈酸性,不利于成骨[19],后期也需要進一步研究以解決這一問題。對于微囊釋放BMP-2 來促進成骨分化的最適相對分子質量,也仍需比較更多不同相對分子質量的微囊以得出更準確的結論。本研究結果為進一步開發可控性骨組織工程技術材料提供了思路。
利益沖突聲明:作者聲明本文無利益沖突。
[參考文獻]
[1] Ball JR, Shelby T, Hernandez F, et al. Delivery of growth
factors to enhance bone repair[J]. Bioengineering (Basel),
2023, 10(11): 1252.
[2] Wei J, Xia X, Xiao S, et al. Sequential dual-biofactor release
from the scaffold of mesoporous HA microspheres
and PLGA matrix for boosting endogenous bone regeneration[
J]. Adv Healthc Mater, 2023, 12(20): e2300624.
[3] Minardi S, Fernandez-Moure JS, Fan D, et al. Biocompatible
PLGA-mesoporous silicon microspheres for the
controlled release of BMP-2 for bone augmentation[J].
Pharmaceutics, 2020, 12(2): 118.
[4] De Witte TM, Fratila-Apachitei LE, Zadpoor AA, et al.
Bone tissue engineering via growth factor delivery: from
scaffolds to complex matrices[J]. Regen Biomater, 2018,
5(4): 197-211.
[5] Guo X, Song P, Li F, et al. Research progress of design
drugs and composite biomaterials in bone tissue engineering[
J]. Int J Nanomedicine, 2023, 18: 3595-3622.
[6] Shah NJ, Macdonald ML, Beben YM, et al. Tunable dual
growth factor delivery from polyelectrolyte multilayer
films[J]. Biomaterials, 2011, 32(26): 6183-6193.
[7] Minardi S, Pandolfi L, Taraballi F, et al. PLGA-mesoporous
silicon microspheres for the in vivo controlled temporospatial
delivery of proteins[J]. ACS Appl Mater Interfaces,
2015, 7(30): 16364-16373.
[8] Jin S, Xia X, Huang J, et al. Recent advances in PLGAbased
biomaterials for bone tissue regeneration[J]. Acta
Biomater, 2021, 127: 56-79.
[9] Sun F, Sun X, Wang H, et al. Application of 3D-printed,
PLGA-based scaffolds in bone tissue engineering[J]. Int
J Mol Sci, 2022, 23(10): 5831.
[10] Wang Y, Zhao L, Zhou L, et al. Sequential release of vascular
endothelial growth factor ?A and bone morphogenetic
protein-2 from osteogenic scaffolds assembled by
PLGA microcapsules: a preliminary study in vitro[J]. Int
J Biol Macromol, 2023, 232: 123330.
[11] 王瑩, 陳晨, 陳剛. BMP-2 緩釋型PLGA微囊作為引導
骨再生支架的初步研究[J]. 南京醫科大學學報(自然科
學版), 2022, 42(9): 1216-1222.
Wang Y, Chen C, Chen G. Preliminary study of a BMP-
2 releasing scaffold comprised of PLGA microspheres
for guiding bone regeneration[J]. J Nanjing Med Univ
(Nat Sci), 2022, 42(9): 1216-1222.
[12] Nyberg E, Holmes C, Witham T, et al. Growth factoreluting
technologies for bone tissue engineering[J]. Drug
Deliv Transl Res, 2016, 6(2): 184-194.
[13] Wani TU, Khan RS, Rather AH, et al. Local dual delivery
therapeutic strategies: using biomaterials for advanced
bone tissue regeneration[J]. J Control Release, 2021,
339: 143-155.
[14] Halloran D, Durbano HW, Nohe A. Bone morphogenetic
protein-2 in development and bone homeostasis[J]. J
Dev Biol, 2020, 8(3): 19.
[15] Shi J, Dai W, Gupta A, et al. Frontiers of hydroxyapatite
composites in bionic bone tissue engineering[J]. Materials
(Basel), 2022, 15(23): 8475.
[16] Rmaidi A, Zelzer M, Sindji L, et al. Impact of the physico-
chemical properties of polymeric microspheres functionalized
with cell adhesion molecules on the behavior
of mesenchymal stromal cells[J]. Mater Sci Eng C Mater
Biol Appl, 2021, 121: 111852.
[17] Zhao D, Zhu T, Li J, et al. Poly(lactic-co-glycolic acid)-
based composite bone-substitute materials[J]. Bioact Mater,
2020, 6(2): 346-360.
[18] Lee SK, Han CM, Park W, et al. Synergistically enhanced
osteoconductivity and anti-inflammation of PLGA/
β -TCP/Mg(OH)2 composite for orthopedic applications
[J]. Mater Sci Eng C Mater Biol Appl, 2019, 94: 65-75.
[19] Yuan Y, Xu Y, Mao Y, et al. Three birds, one stone: an osteo-
microenvironment stage-regulative scaffold for bone
defect repair through modulating early osteo-immunomodulation,
middle neovascularization, and later osteogenesis[
J]. Adv Sci (Weinh), 2024, 11(6): e2306428.
(本文編輯 杜冰)
