厭氧環境下黏土礦物結構Fe(Ⅲ)還原對Cd(Ⅱ)固持特性的影響
2024-11-09 00:00:00范小妹董易坤吳聰王帥廖文娟周衛軍崔浩杰
農業環境科學學報 2024年5期
關鍵詞:黏土礦物;結構態鐵;還原;鎘;吸附
鎘(Cd)作為國際公認的優先控制環境污染物之一,對人類健康危害嚴重。Cd進入人體后,主要富集在腎臟中,并且可以導致慢性Cd中毒,損害腎臟功能。此外,慢性Cd中毒還可能導致骨骼系統的損害和增加心血管疾病的風險,如高血壓和心臟病。Cd污染主要是通過廢水排放、大氣沉降等途徑進入水體和土壤,嚴重危害環境健康。研究表明,Cd污染可顯著降低典型沉水植物苦草植株鮮質量、株高、葉綠素含量和抗逆能力。Cd脅迫引起玉米幼苗葉片萎蔫,單株干質量、葉綠素含量、凈光合速率、蒸騰速率和氣孔導度急劇降低。羅路云等發現,Cd污染降低了稻田土壤真菌群落a多樣性并改變了土壤真菌群落結構。湖南是我國水稻主產區,水稻播種面積和產量均為全國第一,在國家糧食安全戰略中具有重要地位。然而,區域內土壤Cd污染導致稻米Cd(Ⅱ)含量超標問題突出,使得該區域稻田成為目前國內最受關注的環境污染管控地區,嚴重威脅國家糧食安全,也影響著社會發展。因此,Cd(Ⅱ)在稻田土壤一水稻系統的遷移轉化過程與調控備受關注。
土壤組分固持是土壤一水稻系統Cd(Ⅱ)遷移轉化的重要環節,決定著稻田土壤中Cd(Ⅱ)的形態分布及其生物有效性。黏土礦物作為土壤主要固相組分在調控Cd(Ⅱ)有效性方面具有重要作用,兩者的相互作用與礦物類型和結構特性密切相關。自然形成的黏土礦物晶體結構中一般含有不同數量的鐵,這種結構態鐵約占土壤和沉積物總鐵的50%。稻田淹水形成的厭氧環境中廣泛存在的Fe(Ⅱ),還原性有機質,含巰基小分子化合物以及鐵、硫還原菌等,均可還原黏土礦物晶體結構中的Fe(Ⅲ)。結構Fe(Ⅲ)還原過程中會發生原子遷移與結構重排、礦物轉化、礦物溶解與再吸附或再沉淀等一系列反應,改變礦物晶體化學特性。如富鐵蒙皂石還原過程中,結構中二八面體Fe(Ⅲ)還原形成三八面體Fe(Ⅱ)和空位,同時Fe原子從順式八面體位置遷移至反式八面體位置,而微生物還原蒙皂石結構Fe(Ⅲ)時,層間會發生塌陷,誘導“伊利石化”反應,促進蒙皂石向伊利石轉變。此外,黏土礦物結構Fe(Ⅲ)還原時,部分礦物溶解釋放Fe(Ⅱ)和Al(Ⅲ),其中部分釋放的Fe(Ⅱ)會在礦物邊面位點被重新吸附,進入八面體層反式構型的空位中,而釋放的Al(Ⅲ)經過再沉淀反應,可在礦物基面形成新的富含活性Al(Ⅲ)層。上述反應過程中形成的不同形態結構和表面Fe(Ⅱ)以及活性Al(Ⅲ)層,顯著提高了礦物的界面化學反應活性。如還原性富鐵綠脫石邊面位點形成的Fe(Ⅲ)-O-Fe(Ⅱ)可在厭氧條件下氧化As(Ⅲ),且對As(Ⅲ)的氧化能力隨礦物結構中Fe(Ⅲ)還原程度的增加而提高。而在微生物還原過程中礦物基面上形成的活性Al(Ⅲ)層,顯著提高了礦物對有機質的固持性能。然而,有關厭氧環境下黏土礦物結構Fe(Ⅲ)還原如何影響它們與Cd(Ⅱ)的作用特性目前尚不清楚。
本試驗以合成含鐵綠脫石為研究對象,通過化學還原,探討了厭氧條件下綠脫石結構態Fe(Ⅲ)還原對Cd(Ⅱ)吸附和固持性能的影響,揭示了黏土礦物結構Fe(Ⅲ)還原抑制Cd(Ⅱ)吸附的作用機制,結果可為深入理解厭氧環境條件下含鐵黏土礦物與重金屬元素界面化學過程提供依據。
1材料與方法
1.1綠脫石的制備
綠脫石參照Ilgen等的方法制備。稱取5.767gH4Si04和12.0g NaOH加入去離子水混合,用純水稀釋定容至1L,在磁力攪拌器上攪拌過夜直到澄清,獲得試劑A。在合成反應前制備試劑B:將2.0gNa2S204溶于25mL無氧去離子水中,加入1.877g FeS04.7H20混合,用去氧水定容至50mL,然后將120mL試劑A與40mL試劑B倒入200mL反應釜中,密閉搖勻。將反應釜置于150℃烘箱中反應50h。待反應釜冷卻至室溫后,用去離子水將反應產物從反應釜中洗出,離心分離,倒掉上清液,然后用去離子水洗滌3次。將產物轉移到1mol·L-1 NaCI溶液中,置于磁力攪拌器上攪拌分散12h。將產物離心分離,用去離子水洗滌5次去除殘留鹽。清洗后的樣品用草酸銨一草酸溶液洗滌,去除潛在的氧化鐵雜質。草酸鹽試劑由700mL 0.2mol·L-1草酸銨溶液和535mL 0.2mol·L-1草酸溶液組合而成。草酸銨一草酸溶液試劑的pH值為(3.0±0.1)。每0.25g固體樣品加入10mL草酸試劑,放在(25±1)℃的搖床中,避光250r·min-1振蕩4h。然后離心分離固體樣品,加入去離子水沖洗5次,最后將樣品置于凍干機中凍干,研磨過100目篩后,置于厭氧手套箱內保存備用。
1.2還原前后礦物懸濁液制備
未還原礦物懸濁液制備:稱取4.0g過篩綠脫石加入去離子水定容至200mL,獲得20g·L-1未還原合成綠脫石礦物樣品懸濁液,通入N2 30min后轉移至厭氧手套箱內,在手套箱內向懸濁液中投入一枚磁子,密封備用,每次取用時需提前置于磁力攪拌器上攪拌分散均勻。
還原礦物懸濁液制備:參考Liao等的方法,采用Na2S204對綠脫石進行化學還原。取8個100mL厭氧瓶,每個裝入0.56g過篩綠脫石樣品,再分別加入0.5g Na2S2O4,將厭氧瓶移入厭氧手套箱繼續加入40mL無氧去離子水和15mL無氧碳酸氫鈉一檸檬酸鈉混合緩沖溶液(1mol·L-1碳酸氫鈉和0.9mol·L-1檸檬酸鈉溶液按24:1的體積比混合制得)后厭氧密封。將8個厭氧瓶移出厭氧手套箱置于70℃水浴鍋中加熱4h,冷卻,離心分離得到固體樣品后,先用pH 4.0的2mol·L-1 NaCl溶液清洗活化,再分別用pH 7.0的1.0、0.1、0.05mol·L-1 NaCl各清洗一次,最后用無氧去離子水清洗3次,收集全部固體定容至200mL,獲得20g·L-1還原態合成綠脫石礦物樣品懸濁液,通入N230min后轉移至厭氧手套箱內,放人一枚磁子保存備用,取用時提前置于磁力攪拌器上攪拌分散均勻。
1.3還原前后黏土礦物結構中鐵化學形態分析
在手套箱中取1mL20g·L-1礦物懸濁液,加入8mL5mol·L-1 H2S04和幾滴HF將礦物消解,定容至10mL。用鄰啡羅啉比色法測定礦物中Fe(Ⅱ)含量,添加濃度為10%的鹽酸羥胺溶液將Fe(Ⅲ)還原,用鄰啡羅啉比色法測定礦物中總Fe含量,用總Fe含量減去Fe(Ⅱ)含量即可得到Fe(Ⅲ)含量。
在手套箱中取1mL20g·L-1礦物懸濁液,離心分離后轉入手套箱,倒掉上清液,加入10mL 1 mol·L-1NaH2P04溶液(pH 5,去氧),密封后從手套箱取出,置于(25±1)℃搖床中,以250r·min-1轉速,避光振蕩18h,離心分離,取上清液用0.22um濾頭過濾,用鄰啡羅啉比色測定礦物吸附態Fe(Ⅱ)含量,添加濃度為10%的鹽酸羥胺溶液將Fe(Ⅲ)還原后用鄰啡羅啉比色法測定礦物吸附態總Fe含量,用礦物吸附態總Fe含量減去吸附態Fe(Ⅱ)含量即可得到吸附態Fe(Ⅲ)含量。
將測得的礦物中Fe(Ⅱ)和Fe(Ⅲ)含量減去吸附態Fe(Ⅱ)和Fe(Ⅲ)含量,得到礦物結構態Fe(Ⅱ)和Fe(Ⅲ)含量。
1.4還原前后黏土礦物樣品表征
X-射線衍射(XRD)分析:在厭氧手套箱中將還原前后礦物懸濁液均勻涂在干燥潔凈的玻璃片上,于厭氧手套箱中晾干后,進行XRD(型號:XRD6100,島津)分析。測試條件為:Cu Ka輻射(A=0.154 1nm),電壓40 kV,電流30 mA,掃描速度為8°·min-1,掃描范圍為10°~80°。
掃描電子顯微鏡(SEM)分析:在厭氧手套箱內用抽濾機將還原前后樣品固體收集于濾膜上,于厭氧手套箱中晾干、研磨、過篩。樣品測試前在真空干燥箱80℃下處理48h,取少量樣品粘于導電膠帶上并真空鍍金110s,樣品在掃描電鏡(型號:S4800,日立)下進行形貌觀察及能譜分析,加速電壓為0.5~30.0kV。
比表面積(BET)分析:通過氣體吸附法在比表面積及孔徑分析儀(SSA-4000,北京彼奧德電子)上分析。稱取一定質量的樣品,對樣品進行預處理,在105℃下真空加熱2h,在液氮飽和溫度下測定樣品的等溫吸附和脫附,分析樣品比表面積(SSA)和孔徑(PV)分布規律。
X-射線光電子能譜(XPS)分析:在XPS(Quantum 2000,美國pHI)上采集樣品全譜和Fe、0、Cd等元素窄區譜。以A1 Ka為激發光源,X射線強度為35W,分析壓力為5X10-8 Pa。
1.5還原前后黏土礦物吸附Cd(Ⅱ)
礦物吸附平衡試驗:反應中礦物濃度為1g·L-1,Cd(Ⅱ)濃度為30mg·L-1,NaCl濃度為10mmol·L-1,初始pH為6.0,反應時間24h。在此基礎上分別調整Cd(Ⅱ)濃度為2-100mg·L-1,初始pH值為5.0、6.0、7.0和8.0,NaCI濃度為0、10、50、100mmol·L-1。
吸附動力學實驗:實驗溫度為25℃,反應初始pH為6,反應體積為60mL,反應中礦物濃度為1g·L-1,Cd(Ⅱ)濃度為30mg·L-1,NaCI濃度為10mmol·L-1,按時間點(5min、30min、1h、2h、4h、8h、16h和24h)依次取2.5mL的反應液體,用0.22um濾頭搭配注射器過濾,得到清液用于測定不同時間Cd(Ⅱ)的剩余濃度,并計算吸附量。探討還原前后礦物對Cd(Ⅱ)的等溫吸附和吸附動力學以及pH和離子強度對Cd(Ⅱ)吸附的影響。各吸附試驗均設置3組平行,結果取平均值。
1.6數據處理
使用Excel和Origin對數據進行處理并做圖分析,利用Langmuir和Freundlich模型分別對還原前后礦物吸附Cd(Ⅱ)的等溫實驗數據進行擬合。Lang-muir模型方程為:
2結果與討論
2.1還原前后礦物表征
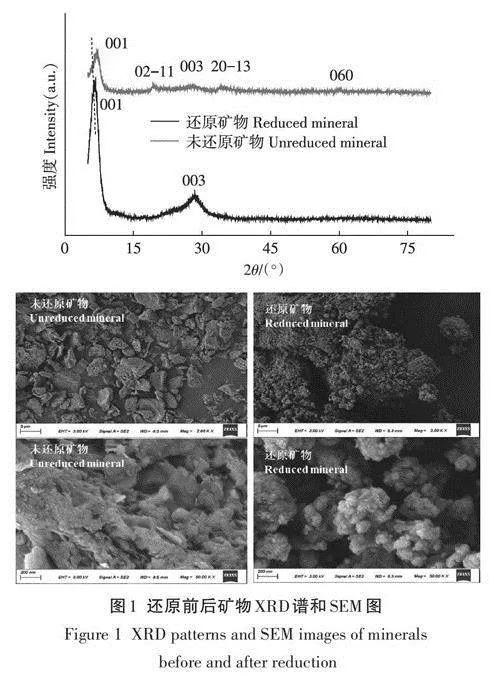
2.1.1礦物XRD和SEM分析
還原前后綠脫石礦物樣品XRD和SEM分析結果如圖l所示。XRD譜中未還原樣品在2為7.07°、19.2°、28.1°、34.2°和60.3°處出現了特征衍射峰,分別為綠脫石001、02-11、003、20-13和060晶面特征衍射峰(PDF#29-1497),這與Ilgen等報道的結果一致。合成綠脫石衍射峰強度較弱,說明合成礦物結晶程度較低。與未還原樣品相比,還原后綠脫石礦物樣品部分特征衍射峰消失,且在001和003處的衍射峰增強,這可能與還原過程中礦物發生結構重排以及顆粒較小礦物的溶解有關。此外,還原后綠脫石001衍射峰向左偏移,d值變大,說明礦物結構層間距變大,這可能是由于礦物還原后部分Fe(Ⅱ)和Fe(Ⅲ)溶出嵌入到礦物結構層中使得層間距變大。在不同放大倍數下,未還原樣品呈現致密塊狀結構或聚集的片狀和塊狀的形貌,而還原樣品則呈現比較松散的顆粒結構,表明還原樣品分散程度高于未還原樣品。
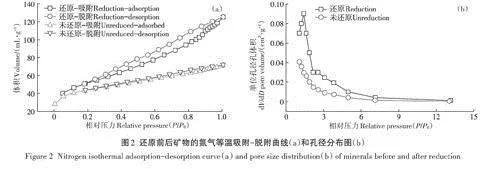
2.1.2礦物比表面積和孔徑分析
還原前后礦物的氮氣等溫吸附一脫附曲線和孔徑分布見圖2。還原和未還原礦物等溫吸附一脫附曲線均為Ⅳ型(根據IUPAC分類),等溫線吸附分支與脫附分支不一致,能觀察到遲滯回線,遲滯環為H4型。未還原礦物多點BET比表面積為151.6m2·g-1,還原處理后礦物的比表面積增加為184.0m2·g-1,比未還原礦物樣品比表面積增加21.4%,這與還原前后礦物SEM分析結果一致。還原前后礦物孔徑分布圖表明,還原前后礦物孔徑分布主要范圍為1.0~8.0nm,其中微孔(lt;2.0nm)所占比例較大,與未還原礦物相比,礦物還原以后微孔比例增大。未還原樣品單點平均孔半徑為1.47nm,還原后樣品的單點平均孔半徑增加為2.12nm。上述結果表明還原處理改變了綠脫石礦物樣品的孔隙結構。
2.1.3還原前后礦物中鐵形態變化特性
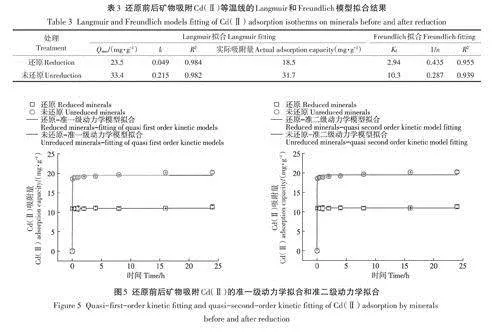
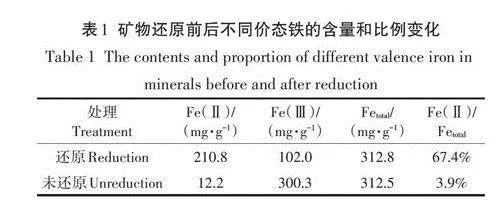
由表1可知,未還原礦物中Fe(Ⅱ)含量為12.2mg·g-1,總Fe含量為312.5mg·g-1,Fe(Ⅱ)/Fe為3.9%。還原礦物中Fe(Ⅱ)含量為210.8mg·g-1,總Fe含量為312.8mg·g-1,Fe(Ⅱ)/Fe為67.4%。結果表明化學還原可顯著提高綠脫石礦物中Fe(Ⅱ)含量。
由表2可知,還原前后礦物樣品中結構態Fe占比最大,未還原礦物中結構態Fe占比為98.6%,其中結構態Fe(Ⅱ)和Fe(Ⅲ)分別為總Fe的3.9%和94.7%。未還原礦物中吸附態Fe占總Fe的1.4%,其中吸附態Fe(Ⅲ)占總Fe的1.4%,而吸附態Fe(Ⅱ)幾乎可以忽略(0.01%)。還原礦物中結構態Fe占比為84.8%,其中結構態Fe(Ⅱ)和Fe(Ⅲ)分別為總Fe的57.9%和26.6%。還原礦物中吸附態Fe占總Fe的15.5%.其中吸附態Fe(Ⅱ)占總鐵的9.5%,吸附態Fe(Ⅲ)占總Fe的6.0%。結果表明礦物經過還原后,吸附態Fe(Ⅱ)和結構態Fe(Ⅱ)的比例會同時增加。
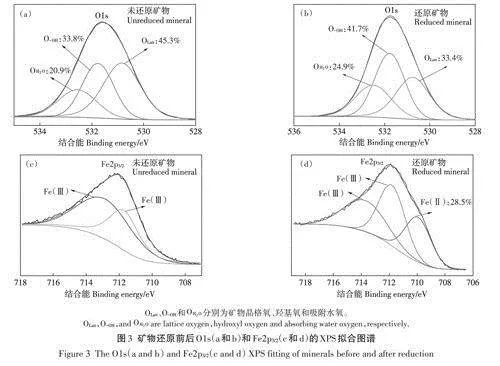
2.1.4XPS分析
還原前后礦物的Ols XPS窄區譜擬合結果見圖3a和圖3b。3個峰結合能值分別為530.9、531.9、532.9eV,分別代表品格氧、羥基氧和吸附水氧。未還原礦物的品格氧、羥基氧和吸附水氧的占比分別為45.3%、33.8%和20.9%,而還原礦物的品格氧、羥基氧和吸附水氧的占比分別為33.4%、41.7%和24.9%。可見,礦物還原后品格氧降低了11.9個百分點,羥基氧占比增加了7.9個百分點,說明還原處理增加了礦物表面的羥基基團數量。未還原和還原礦物的Fe2p3i2 XPS圖譜擬合結果如圖3c和圖3d,未還原樣品中未測出Fe(Ⅱ),而還原樣品中Fe(Ⅱ)/Fe為28.5%,還原前后礦物Fe(Ⅱ)含量均低于酸溶法結果(67.4%和3.9%)。這可能是因為酸溶法測得的是礦物中全部Fe(Ⅱ),XPS測得的是礦物近表面Fe(Ⅱ),而近表面Fe(Ⅱ)會優先被氧化,所以Fe(Ⅱ)/Fe百分比含量偏低。
2.2還原前后礦物吸附固持Cd(Ⅱ)特性
2.2.1還原前后綠脫石對Cd(Ⅱ)的等溫吸附
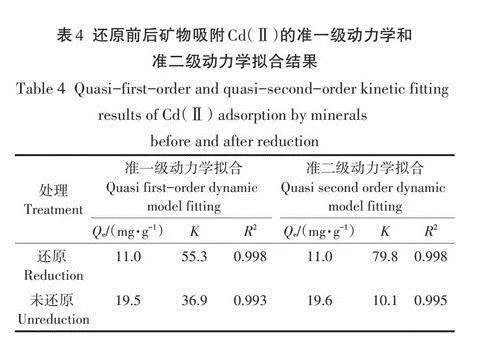
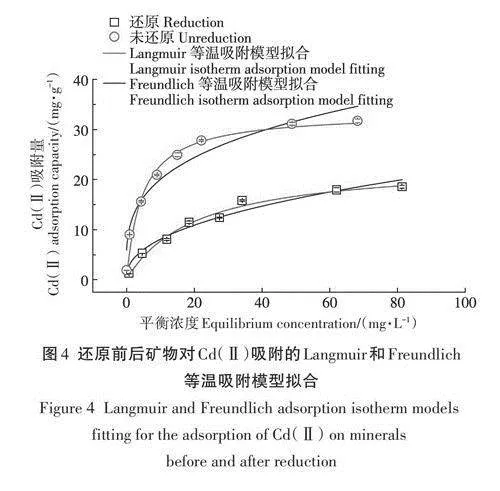
還原與未還原礦物對Cd(Ⅱ)的等溫吸附結果如圖4所示,在同等條件下,未還原綠脫石礦物對Cd(Ⅱ)的吸附量大于還原礦物,還原前后綠脫石對Cd(Ⅱ)的吸附量都與Cd(Ⅱ)平衡濃度呈現正相關。當Cd(Ⅱ)平衡濃度lt;20mg·L-1時,還原前后綠脫石對Cd(Ⅱ)吸附量增加的速率較快;當平衡濃度gt;20mg·L-1時,吸附量增加趨于平緩并最終達到吸附平衡。
由表3可見,Langmuir等溫吸附模型擬合結果的R2值更高,說明Langmuir模型方程相較于Freundlich模型方程更適合擬合綠脫石礦物對Cd(Ⅱ)的吸附實驗數據,Langmuir模型和Freundlich模型分別描述了均勻表面和非均質表面上的吸附情況,結果表明不同還原比的綠脫石礦物對Cd(Ⅱ)的吸附是單分子型。實際測得在Cd(Ⅱ)初始濃度為100mg·L-1時還原和未還原礦物對Cd(Ⅱ)的最大吸附量分別為18.5mg·g-1和31.7mg·g-1,未還原礦物吸附量約為還原礦物的1.71倍。利用Langmuir等溫吸附模型擬合計算出還原和未還原礦物對Cd(Ⅱ)的最大吸附量分別為23.5mg·g-1和33.4mg·g-1,其中還原礦物擬合最大吸附量高于實際最大吸附量,這與本研究中初始Cd(Ⅱ)濃度范圍內(0-100mg·L-1)礦物對Cd(Ⅱ)的吸附沒有完全達到飽和有關。實驗Cd(Ⅱ)濃度范圍內的未還原礦物對Cd(Ⅱ)的最大吸附量約為還原礦物的1.42倍。
2.2.2還原前后綠脫石吸附Cd(Ⅱ)的動力學
在pH為6.0、室溫25℃、反應體積10.0mL、礦物樣品0.01g、Cd(Ⅱ)濃度為30.0mg·L-1、NaCI濃度為10.0mmol·L-1的條件下,對還原和未還原礦物進行吸附Cd(Ⅱ)不同時間的動力學過程,并分別進行了準一級動力學和準二級動力學模型擬合(圖5)。可以看出還原和未還原礦物吸附Cd(Ⅱ)的速率很快,還原前后礦物在反應5min時的吸附量就已接近最大吸附量,隨后吸附量增加速率緩慢,最終達到吸附平衡。還原礦物在24h時的吸附量為11.4mg·g-1,在5min時的吸附量為10.9mg·g-1,超過95%的Cd(Ⅱ)在5min內被吸附。未還原礦物在24h時的吸附量為20.3mg·g-1,在5min時的吸附量為18.6mg·g-1,超過91%的Cd(Ⅱ)在5 min內被吸附。
由表4可見,準一級動力學和準二級動力學模型的R2值均較高,且兩種模型擬合計算的平衡吸附量Q。十分接近,但準二級動力學模型的R2值略高于準一級動力學。準一級動力學模型表示吸附劑對吸附質的結合位點較少,吸附主要受擴散作用控制;準二級動力學模型通常與化學反應控制整體吸附動力學有關,吸附劑表面有多個用于吸附的交換位點。上述分析表明還原前后礦物對Cd(Ⅱ)的吸附過程以化學吸附為主,這與Cd(Ⅱ)離子可通過礦物表面羥基絡合以及層間離子交換等進行吸附作用有關。
2.2.3 pH對還原前后綠脫石吸附Cd(Ⅱ)的影響
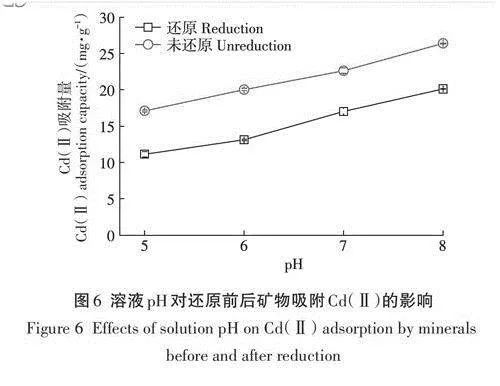
在室溫25℃、反應體積10.0mL、礦物樣品0.01g、Cd(Ⅱ)濃度30.0mg·L-1、NaCl濃度10mmol·L-1的條件下,探究不同溶液pH(5.0-8.0)對還原前后礦物吸附Cd(Ⅱ)的影響,結果如圖6所示,還原前后礦物對Cd(Ⅱ)的吸附量隨著pH的增大而增大。在pH5.0時,還原礦物對Cd(Ⅱ)的吸附量為11.1mg·g-1,未還原礦物對Cd(Ⅱ)的吸附量為17.1mg·g-1。在pH8.0時,還原礦物對Cd(Ⅱ)的吸附量為20.1mg·g-1,吸附率約67.0%,未還原礦物對Cd(Ⅱ)的吸附量為26.4mg·g-1,吸附率約87.9%。這說明反應初始pH會影響還原前后的礦物對Cd(Ⅱ)的吸附量,且隨著pH的升高,礦物對Cd(Ⅱ)的吸附率也會增加。已有研究表明,酸性條件下,溶液的pH越低,H+濃度越高,其會占據黏土礦物的吸附點位,不利于黏土礦物對Cd(Ⅱ)的吸附。而隨著pH的升高,H+濃度降低,黏土礦物表面負電荷密度相應增加,增強了對Cd(Ⅱ)的靜電引力,且OH-濃度的增加使得Cd(Ⅱ)與溶液中的OH-發生沉淀作用而吸附在黏土礦物表面,在中性或是弱堿性條件下,黏土礦物對Cd(Ⅱ)的吸附明顯增強。
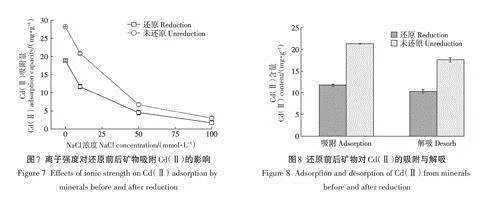
2.2.4離子強度對還原前后綠脫石吸附Cd(Ⅱ)的影響
在pH為6.0、室溫25℃、反應體積10.0mL、礦物樣品0.01g、Cd(Ⅱ)濃度30.0mg·L-1的條件下,探究溶液中NaCl離子強度對還原前后礦物吸附Cd(Ⅱ)的影響(圖7)。由圖可見,當無NaCl加入時,還原前后礦物對Cd(Ⅱ)的吸附量達到最大,分別為28.2mg·g-1和18.9mg·g-1,溶液中Cd(Ⅱ)的去除率分別為94.1%和63.0%。當溶液中NaCI濃度逐漸增加后,還原前后礦物對Cd(Ⅱ)的吸附量均相應降低,當溶液中NaCI濃度增加至100mmol·L-1時,還原礦物對Cd(Ⅱ)的吸附量降至1.69mg·g-1,溶液中Cd(Ⅱ)的去除率為5.63%,而未還原礦物對Cd(Ⅱ)的吸附量為2.96mg·g-1,溶液中Cd(Ⅱ)的去除率為9.87%。與未添加Na-CI相比,還原前后礦物對溶液中Cd(Ⅱ)的去除率分別下降了84.23%和57.34%,這說明離子強度增大,會對還原前后的礦物吸附Cd(Ⅱ)產生較大的抑制作用,且對未還原礦物吸附Cd(Ⅱ)的抑制作用更大。這是由于溶液中的陽離子會在黏土礦物表面與重金屬離子競爭吸附位點,從而降低黏土礦物對重金屬離子的吸收,且離子濃度和價態越高的陽離子對吸附位點的競爭作用越大,不利于黏土礦物對于Cd(Ⅱ)的吸附。
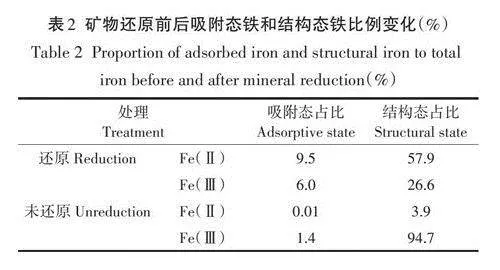
2.2.5還原前后綠脫石吸附Cd(Ⅱ)的解吸
還原前后礦物相同條件下吸附Cd(Ⅱ)后,用pH6.0的0.1mol·L-1去氧CaC12進行解吸,結果見圖8。其中,還原礦物吸附的Cd(Ⅱ)經CaC12解吸后釋放量為10.4mg·g-1,解吸率為87.9%;而未還原礦物吸附的Cd(Ⅱ)解吸量為17.6mg·g-1,解吸率為82.5%。上述分析顯示,還原礦物對Cd(Ⅱ)的吸附量雖然低于未還原礦物,但解吸率略高于未還原礦物,表明綠脫石結構態鐵還原降低了礦物對Cd(Ⅱ)的吸附和固持性能。
2.3還原前后綠脫石吸附Cd(Ⅱ)的機制分析
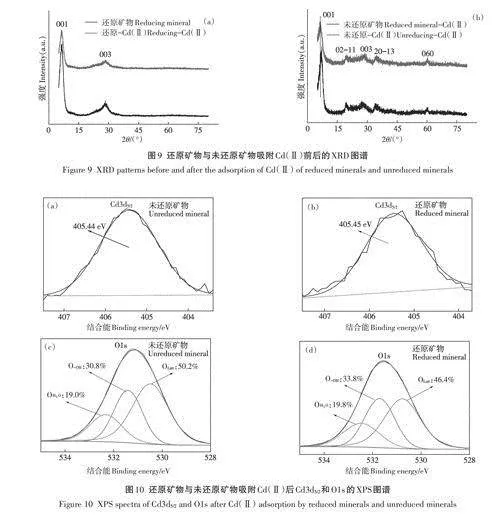
圖9為還原與未還原礦物吸附Cd(Ⅱ)前后的XRD圖。未還原礦物吸附Cd(Ⅱ)后,礦物001晶面特征衍射峰向左偏移,表明吸附過程中Cd(Ⅱ)進入到礦物層間,使得層間距變大。而還原礦物吸附Cd(Ⅱ)前后的XRD特征衍射峰位置幾乎沒有變化,表明還原礦物吸附過程中Cd(Ⅱ)沒有進入層間。已有研究表明,黏土礦物結構Fe(Ⅲ)還原過程中,部分還原形成的Fe(Ⅱ)會進入層間。由于Cd(Ⅱ)的離子半徑(97pm)大于Fe(Ⅱ)離子(61pm),其在礦物層間位點交換競爭能力弱于Fe(Ⅱ),使得吸附過程中Cd(Ⅱ)難以通過離子交換進入結構層間,這可能是綠脫石礦物還原后吸附Cd(Ⅱ)減少的原因之一。
還原前后礦物吸附Cd(Ⅱ)后Cd3d的XPS圖譜結果如圖lOa和圖lOb。吸附后還原和未還原礦物的Cd3d結合能值分別為405.45eV和405.44eV,為Cd(Ⅱ)的結合峰,說明Cd沒有價態的變化,表明Cd(Ⅱ)通過與綠脫石表面羥基配位絡合吸附在了礦物表面。還原前后礦物吸附Cd(Ⅱ)后的Ols XPS圖譜擬合結果如圖lOc和圖lOd。還原礦物吸附Cd(Ⅱ)后的品格氧、羥基氧和吸附水氧的占比分別為46.4%、33.8%和19.8%;未還原礦物吸附Cd(Ⅱ)后的品格氧、羥基氧和吸附水氧的占比分別為50.2%、30.8%和19.0%。兩種礦物吸附Cd(Ⅱ)后的羥基氧占比(33.8%、30.8%)比反應前的羥基氧占比(41.7%、33.8%)更少,說明綠脫石還原前后樣品吸附Cd(Ⅱ)過程中羥基基團均參與了Cd(Ⅱ)的固持。
上述XRD和XPS分析結果表明,還原和未還原礦物吸附Cd(Ⅱ)存在層間嵌入和表面絡合兩種方式。未還原礦物在吸附Cd(Ⅱ)后層間距變大(圖9b),礦物表面羥基比例也由33.8%下降至30.8%,說明層間嵌入和表面絡合兩種方式在未還原礦物吸附Cd(Ⅱ)過程中均存在。礦物結構Fe(Ⅲ)還原過程中,礦物比表面積增加,礦物表面羥基基團比例由33.8%增加至41.7%;而吸附Cd(Ⅱ)后,還原礦物表面羥基比例又下降至33.8%。但還原礦物吸附Cd(Ⅱ)后,礦物層間距幾乎沒有變化(圖9a)。在含鐵黏土礦物化學還原過程中,部分結構Fe(Ⅲ)發生還原溶解、釋放與再吸附,形成的Fe(Ⅱ)部分進入礦物層間,使礦物層間距變大(圖1),另一部分Fe(Ⅱ)會吸附在礦物表面(表2)。另外,由于Cd(Ⅱ)的離子半徑(97 pm)大于Fe(Ⅱ)離子(61 pm),其在礦物表面和層間位點的交換競爭能力弱于Fe(Ⅱ),使得吸附過程中Cd(Ⅱ)難以通過離子交換競爭表面吸附Fe(Ⅱ)的活性位點以及取代層間Fe(Ⅱ)。因此,還原礦物對Cd(Ⅱ)的吸附主要為表面絡合方式,相應也更易于解吸活化(圖8)。綜上,含鐵黏土礦物結構Fe(Ⅲ)還原過程中形成Fe(Ⅱ)進入層間和表面吸附這兩方面作用是還原后礦物吸附Cd(Ⅱ)減少的主要原因,且前者作用可能更大。
3結論
(1)厭氧條件下黏土礦物結構Fe(Ⅲ)還原顯著降低了礦物對Cd(Ⅱ)的吸附和固持性能。
(2)還原前后兩種礦物對Cd(Ⅱ)的吸附量隨著離子強度增加而降低,還原礦物吸附Cd(Ⅱ)的降幅明顯低于未還原礦物。
(3)黏土礦物結構Fe(Ⅲ)還原過程中形成的Fe(Ⅱ)進入層間和表面吸附是還原后礦物吸附Cd(Ⅱ)減少的主要原因。
