腸舌狀絳蟲線粒體基因組特征及系統發育分析
2024-12-31 00:00:00陳秀琴邱陽元呂慶博黃梅清
畜牧獸醫學報 2024年12期



摘" 要: 本研究旨在獲得腸舌狀絳蟲線粒體基因組全序列,了解其序列的結構特征,探究腸舌狀絳蟲系統發育相關信息。從麥穗魚中獲得腸舌狀絳蟲,提取基因組DNA,通過Illumina測序技術對腸舌狀絳蟲的線粒體全基因組進行測序、組裝和注釋,并進行生物信息學分析;從GenBank數據庫中下載8個科34種絳蟲的線粒體基因組序列,運用最大似然法和貝葉斯法構建系統發育樹。結果顯示,腸舌狀絳蟲線粒體基因組全長為13 655 bp,A+T含量為67.6%,其堿基組成具有明顯的AT偏向性。腸舌狀絳蟲線粒體基因組包括12個蛋白編碼基因(protein-coding genes, PCGs)、22個tRNA基因、2個rRNA基因和2個非編碼控制區。在12個PCGs中,除cox3基因使用TTG作為起始密碼子外,其余11個PCGs均使用ATG作為起始密碼子;終止密碼子方面,5個PCGs使用TAA,5個PCGs使用TAG,而cox3基因和nad3基因均沒有終止密碼子。PCGs使用密碼子頻率最高的是UUA,最低的是CGA。22個tRNA基因總長為1 272 bp,大多數tRNA能形成典型的三葉草結構,但是trnS1和trnR由于缺少二氫尿嘧啶臂無法形成典型的三葉草結構。nad2、nad6和nad4基因更適合作為腸舌狀絳蟲的分子標記。系統發育樹結果表明,腸舌狀絳蟲與雙線絳蟲親緣關系最近,并與雙槽頭屬絳蟲(Dibothriocephalus)形成姐妹群。本研究獲得了腸舌狀絳蟲的線粒體全基因組,為研究腸舌狀絳蟲的分類學和系統學提供參考。
關鍵詞: 腸舌狀絳蟲;線粒體基因組;結構特征;系統發育
中圖分類號: S852.734
文獻標志碼:A
文章編號:0366-6964(2024)12-5725-13
doi: 10.11843/j.issn.0366-6964.2024.12.035
開放科學(資源服務)標識碼(OSID):
收稿日期:2024-02-02
基金項目:福建省屬公益類科研院所項目(2023R1024002);福建省農業科學院自由探索科技創新項目(ZYTS2023017)
作者簡介:陳秀琴(1988-),女,福建漳州人,助理研究員,博士,主要從事畜禽疫病診斷方法研究,E-mail: lyunxqchen@163.com
*通信作者:黃梅清,主要從事畜禽疫病診斷方法研究,E-mail: meiqingmail@126.com
Characteristics and Phylogenetic Analysis of Mitochondrial Genome in the Ligula intestinalis
CHEN" Xiuqin1, QIU" Yangyuan2, L Qingbo2, HUANG" Meiqing1*
(1.Institute of Animal Husbandry and Veterinary Medicine, Fujian Academy of Agricultural Scienc/Fujian Animal Diseases Control Technology Development Center, Fuzhou, Fujian 350013," China;
2.Key Laboratory of Zoonosis Research, Ministry of Education/Institute of Zoonosis, College of
Veterinary Medicine, Jilin University, Changchun 130062," China)
Abstract:" This experiment was conducted to obtain the complete mitochondrial genome sequence of Ligula intestinalis, revealed the sequence’s structural features, and explored the phylogenetic information of L. intestinalis. Several L. intestinalis were obtained from Pseudorasbora parva and genomic DNA was extracted. The complete mitochondrial genome of L. intestinalis was sequenced, assembled, and annotated using Illumina sequencing technology after quality control of the DNA, followed by bioinformatic analysis. Maximum likelihood (ML) and bayesian inference (BI) analysis were used to construct the phylogenetic tree. The results showed that the complete mitochondrial genome of L. intestinalis was 13 655 bp in length, and its A+T content was 67.6%, exhibiting a clear AT bias. The complete mitochondrial genome was composed of 12 protein-coding genes (PCGs), 22 tRNA genes, two rRNA genes and two NCRs (non-coding regions). Among the 12 PCGs,11 used ATG as the start codon, except for cox3, which had a TTG initial one. Among the termination codons, five out of twelve were identified as TAA, five as TAG, while neither the cox3 nor nad3 genes had termination codons. The total length of the 22 tRNA genes was 1 272 bp. Most tRNAs have a conventional cloverleaf structure, but trnS1 and trnR lack dihydrouridine arms of tRNA. The nad2, nad6 and nad4 genes are more suitable as molecular markers for L. intestinalis. The results of the phylogenetic tree showed that Ligula and Digramma were most closely related to each other, forming a sister group with Dibothriocephalus. The complete mitochondrial genome of L. intestinalis was obtained. This study will provide a reference for the study of the taxonomy and systematics of L. intestinalis.
Key words: Ligula intestinalis; mitogenome; structure characteristics; phylogeny
*Corresponding author:" HUANG Meiqing,E-mail: meiqingmail@126.com
腸舌狀絳蟲(Ligula intestinalis)是一種寄生于淡水魚的大型絳蟲,主要感染鯉科魚類[1-2]。腸舌狀絳蟲感染會嚴重影響魚的生長性能,降低繁殖力,甚至造成魚類死亡[3]。由于絳蟲裂頭蚴無特征性形態,僅依靠形態特征進行物種鑒定極具挑戰[4]。例如,裂頭科絳蟲中的腸舌狀絳蟲(舌狀屬,Ligula)和雙線絳蟲(雙線屬,Digramma)的裂頭蚴均呈現“面條樣”[4-5]。此外,舌狀屬和雙線屬的分類地位也一直存在爭議。有研究報道舌狀屬和雙線屬可能不是獨立的屬,舌狀屬應屬于雙線屬[6-7]。
盡管使用核基因分子標記的系統發育分析在揭示許多類群的進化關系方面發揮了重要作用,但是核基因分子標記顯示的物種有時變異小或者分支支持有時較低[5]。相比之下,線粒體基因組具有嚴格的母系遺傳、進化速度相對較快、無旁系同源基因和相對保守的基因組結構,是一種理想的分子遺傳標記,已經廣泛應用于物種鑒定、群體遺傳學、分子診斷以及系統發育等研究領域[5,8-9]。盡管線粒體基因適用于多種生物體的系統發育研究,但是單個線粒體基因片段通常較短,難以完全解析不同物種間的進化關系[5,10]。而完整的線粒體基因組序列可顯著提高系統發育分析能力,并且能夠在深層次上準確解析分類學關系。因此,基于線粒體基因組的系統發育已廣泛應用于絳蟲進化發育研究[11-14]。已報道的絳蟲線粒體基因組大多采用長PCR擴增法[11-14],這種方法不僅操作繁瑣,而且由于可供參考的絳蟲線粒體基因組較少,導致引物設計不合適,使得獲得的線粒體基因組信息不準確。近年來,高通量測序技術的發展為獲取線粒體基因組提供了更便捷的工具。然而,該方法尚未應用于腸舌狀絳蟲的線粒體基因組研究。
本研究采用高通量測序技術Illumina平臺對腸舌狀絳蟲的線粒體全基因組進行測序,分析其基因結構、堿基組成、蛋白質編碼基因(protein-coding genes, PCGs)和密碼子使用情況, 結合已發表的絳蟲線粒體全基因組序列對腸舌狀絳蟲進行系統發育分析, 以期為腸舌狀絳蟲的分子鑒定、分類地位和線粒體基因組研究奠定基礎。
1" 材料與方法
1.1" 腸舌狀絳蟲基因組DNA提取
腸舌狀絳蟲分離自黑龍江省扶余市捕撈的麥穗魚,將蟲體剪成小塊置于1.5 mL離心管中,加入裂解液和蛋白酶K溶液,56℃裂解1.5 h。按照基因組DNA提取試劑盒(TIANGEN, Code No.DP304)說明書提取基因組DNA,通過瓊脂糖凝膠電泳檢測基因組DNA的純度。在前期的研究中,通過形態學以及擴增蟲體的線粒體cox1基因鑒定為腸舌狀絳蟲。
1.2" 高通量測序
檢測合格的DNA樣品分別采用Covaris超聲波破碎儀隨機分割成長度為350 bp左右的片段,采用標準的Illumina TruSeq Nano DNA LT文庫制備流程構建腸舌狀絳蟲DNA樣本上機文庫,委托南京派森諾基因科技有限公司,使用Hiseq Xten PE150平臺進行高通量測序。采FSATP對下機數據進行質控,刪除讀段(reads)中的測序接頭以及引物序列,過濾長度低于50 bp、Q值低于20或N含量大于3 的讀段,得到高質量數據(clean data)。
1.3" 線粒體基因組序列拼接和注釋
用軟件SPAdes v3.15.5、IDBA和GetOrganelle v1.7.7.0 完成線粒體基因組序列的組裝。然后, 利用在線注釋工具MITOS2 (http://mitos2.bioinf.uni-leipzig.de/index.py)對線粒體基因組序列進行識別和注釋, 并使用CGView (https://cgview.ca)生成線粒體基因組的基因圖譜。
1.4" 腸舌狀絳蟲mtDNA生物信息學分析
使用PhyloSuite軟件統計腸舌狀絳蟲全基因組及各基因的長度、AT 含量、核苷酸偏倚度、PCGs各基因的起始和終止密碼子,計算相對同義密碼子使用度(relative synonymous codon usage, RSCU)等信息[15]。
1.5" 基因差異位點分析
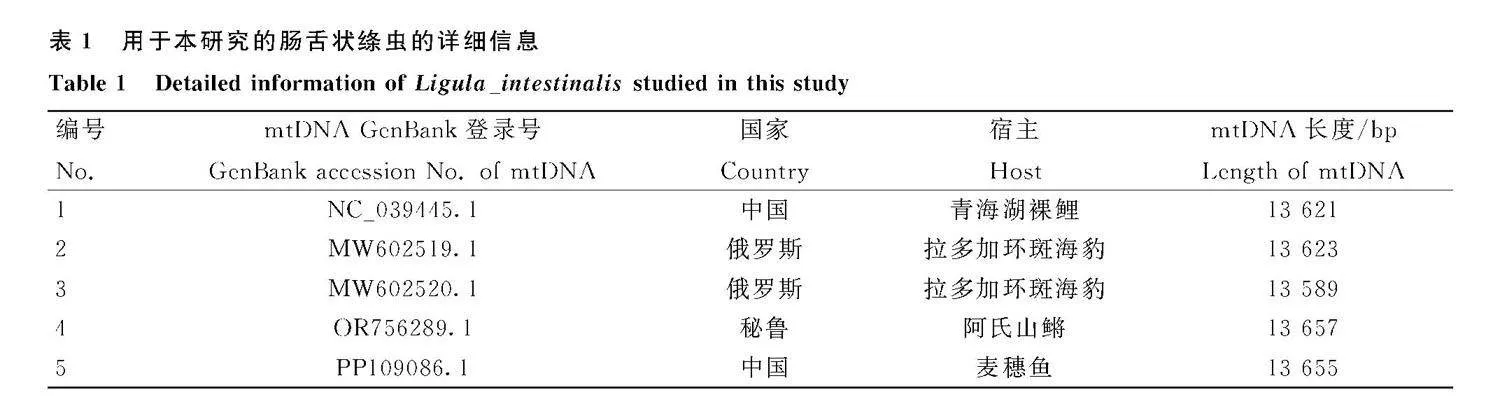
從GenBank數據庫下載所有腸舌狀絳蟲全基因組,運用MEGA 6.0軟件對12個PCGs和2個rRNA 基因進行多重比對,再通過DnaSP 6 軟件分析舌狀絳蟲基因組的基因差異位點。用于基因差異位點分析的腸舌狀絳蟲具體信息見表1。
1.6" 系統發育分析
從GenBank數據庫中下載8個科34種絳蟲的線粒體全基因組作為參考序列,以長尾窄體吸蟲(Lyperosomum longicauda, NC_048467.1)作為外群(表2)。使用PhyloSuite軟件提取所有從GenBank數據庫中下載序列的36個基因信息(12個PCGs、2個rRNAs和22個tRNAs),將PCGs翻譯成氨基酸序列。使用MAFFT軟件和MACSE軟件對34種絳蟲的線粒體基因組12個PCGs進行批量比對。將獲得的比對結果導入Gblocks以消除不明確的序列[16]。根據ModelFinder中最低的貝葉斯信息標準得分選擇最佳核苷酸替代模型[17]。
采用最大似然法(maximum likelihood,ML) 和貝葉斯法(Bayesian inference,BI) 來構建系統發育樹,其中,ML進化樹運用IQ-TREE v1.6.12軟件進行構建[18],系統發育樹的各分支置信水平用自展檢驗(bootstrap test)進行評估,重復抽樣次數為5 000次。BI進化樹則運用MrBayes v3.2.6軟件進行構建[19],運行世代數設置為5×106,運行4條蒙特卡洛馬爾夫鏈(Markov chain Monte Carlo,MCMC),運行每1 000世代取樣1 次。當分列頻率平均標準差(average standard deviation of split frequencies)低于0.01,ESS值(estimated sample size)大于200,以及PSRF值(potential scale reduction factor)接近1時則認為收斂。最后去掉運算開始的前25%取樣,剩下的取樣則用于測算貝葉斯后驗概率(posterior probability,PP)。所得到的進化樹通過在線工具iTOL進行可視化和美化[20]。
2" 結" 果
2.1" 腸舌狀絳蟲線粒體全基因組分析
2.1.1" 線粒體基因組的結構分析
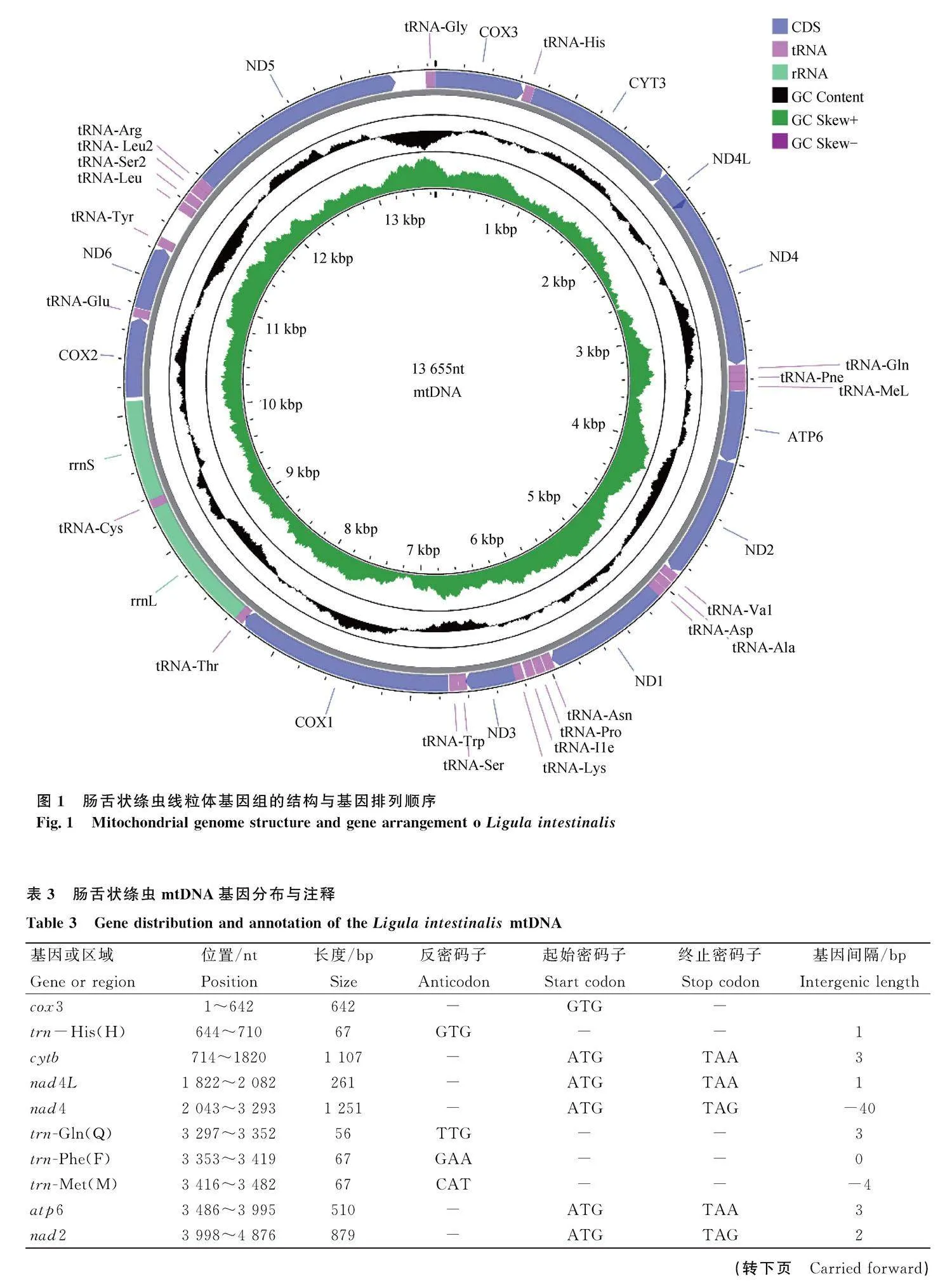
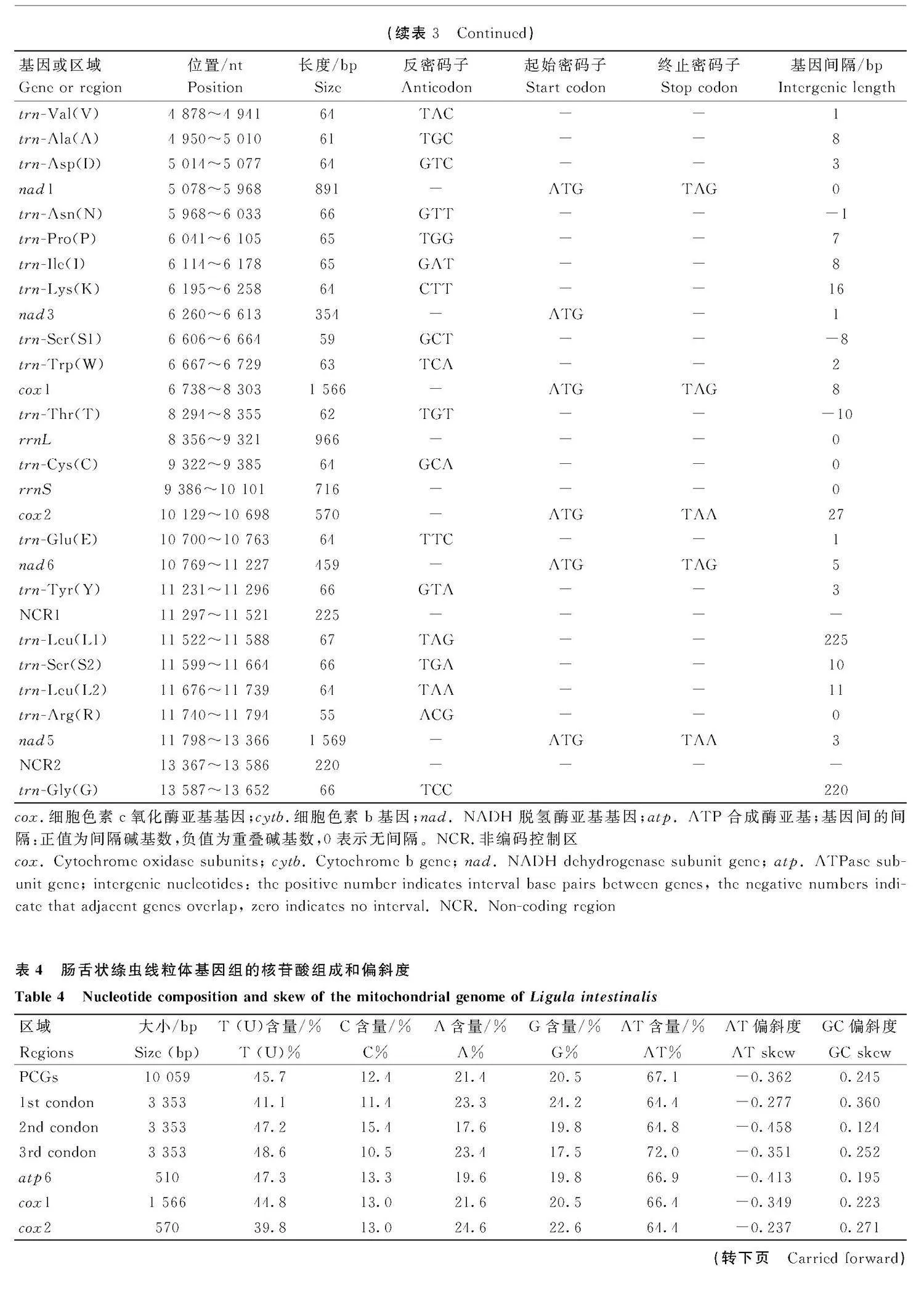
腸舌狀絳蟲的線粒體基因組為典型的閉合雙鏈DNA分子,全長為13 655 bp,共編碼36個基因, 包括12個PCGs(cox3、cytb、nad4L、nad4、atp6、nad2、nad1、nad3、cox1、cox2、nad6、nad5)、22 個tRNAs(trnG、trnH、trnQ等)和2個rRNAs(rrnL和rrnS)和2個非編碼控制區(non-coding region, NCR)(圖1、表3)。
2.1.2" 堿基組成特點
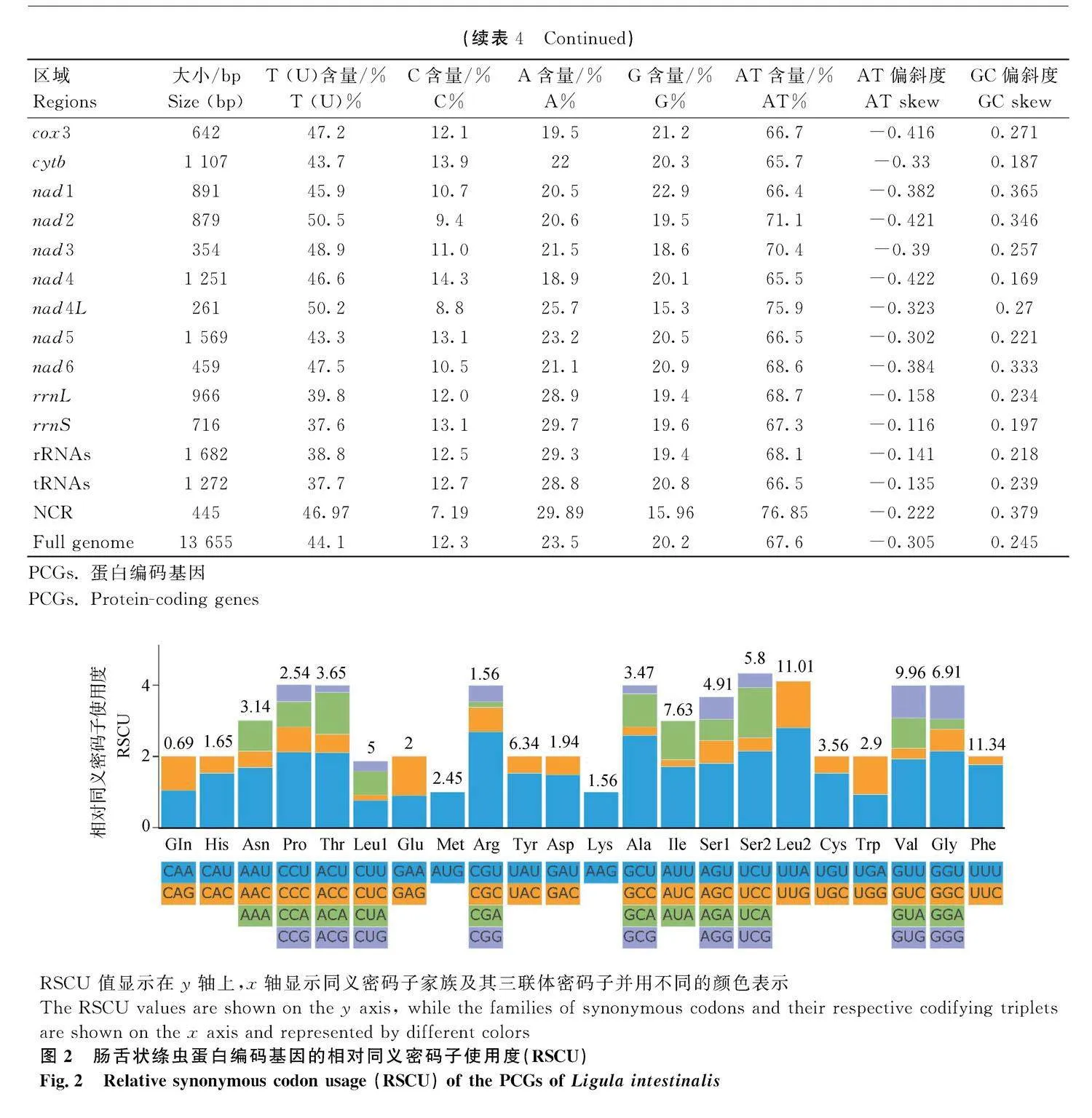
腸舌狀絳蟲的線粒體基因組的A、T、C、G堿基組成分別為23.5%、44.1%、12.3%、20.2%,堿基呈AT偏好, 其A+T含量為67.6%,AT偏斜度為-0.305,GC偏斜度為0.245,同時PCGs、tRNA、rRNA和NCR均呈AT偏好,且線粒體基因組和PCGs的AT偏斜度高于GC偏斜度(表4)。
2.1.3" 蛋白編碼基因及密碼子使用情況
腸舌狀絳蟲所有PCGs的總長度(10 059 bp)占線粒體全基因組總長度的73.67%,各個PCG的長度從261 bp(nad4L)~1 569 bp (nad5) 不等,AT含量在64.4%(cox2) ~71.1%(nad2)之間,呈現明顯的AT偏斜,3 個位置的AT含量均遠高于GC含量,且密碼子第3位的AT含量均明顯高于其它2個位點(表4)。對12個PCGs的起始密碼子和終止密碼子進行分析,結果表明,除了cox3基因使用非標準起始密碼子TTG外,其余11個PCGs均使用ATG作為起始密碼子;終止密碼子方面,5 個PCGs使用TAA,5 個PCGs使用TAG,而cox3基因和nad3基因都沒有終止密碼子(表3)。排除終止密碼子,腸舌狀絳蟲的PCGs共使用3 353個三聯體密碼子。
使用PhyloSuite軟件分析腸舌狀絳蟲線粒體基因組12個PCGs的RSCU,如圖2所示,PCGs使用密碼子頻率最高的是UUA(Leu2,RSCU=2.81),其次是CGU(Arg,RSCU=2.69)和GCU(Ala,RSCU=2.59);PCGs使用密碼子頻率最低的是CGA(Arg,RSCU=0.15),其次是AUC(Ile,RSCU=0.2)和UUC(Phe,RSCU=0.23)。以上結果表明,與使用C或G結尾的密碼子相比,大多數以A或U(T)結尾的密碼子均顯著表達(RSCUgt;1) ,驗證了其PCGs的密碼子存在AT偏好。
2.1.4" tRNA和rRNA
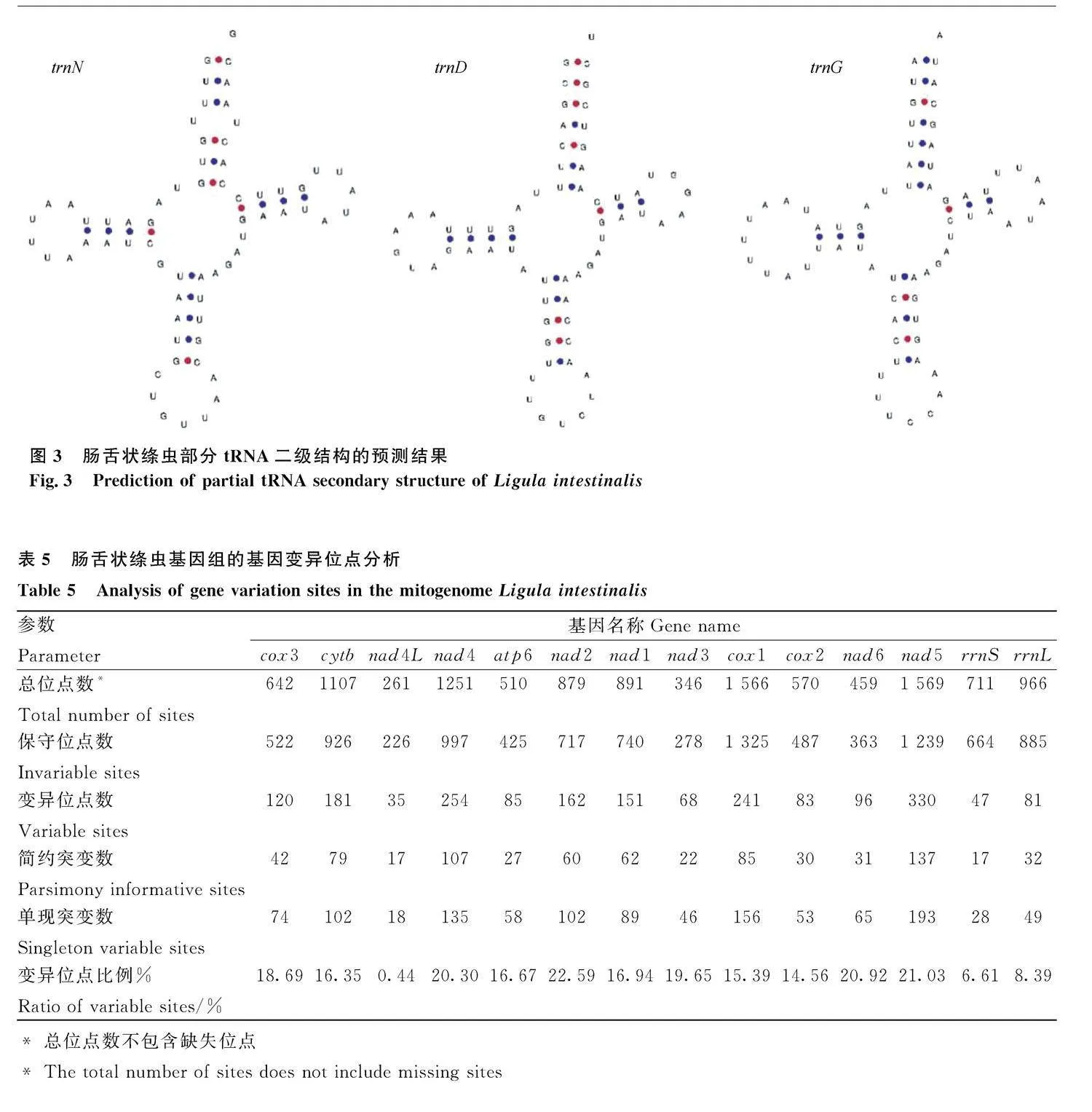
腸舌狀絳蟲線粒體基因組的22個tRNA基因總長為1 272 bp,A+T 的含量為66.5%,tRNA基因的長度在55 bp(trnR) ~67 bp(trnF、trnM和trnL1)之間。多數tRNA都可以折疊成典型的三葉草結構,但是trnS1和trnR由于缺少二氫尿嘧啶臂無法形成典型的三葉草結構。在20個tRNA的二級結構中,氨基酸接受臂、TΨC 臂、反義密碼子臂和二氫尿嘧啶臂都出現了G-U 錯配現象(圖3,僅展示3個)。
腸舌狀絳蟲線粒體基因組的兩個rRNA 基因(rrnL和rrnS)長度分別為966 bp 和716 bp,兩者分別位于trnT 和trnC,trnC和cox2之間,總長度為1 682 bp,A+T 的含量為68.1%(圖1,表4)。
2.1.5" 非編碼控制區
腸舌狀絳蟲線粒體基因組的非編碼控制區有兩個,分別位于trnY和trnL1之間,nad5和trnG之間,總長度為445 bp,占整個線粒體基因組總長度的4.21%,A+T含量為76.85%,比其它基因的A+T含量都更高(圖1、表4)。相比較于線粒體基因組和PCGs,兩個NCR的AT偏斜度小于GC偏斜度(表3)。
2.1.6" 基因重疊和間隔序列
腸舌狀絳蟲線粒體基因組共有基因間隔23處,總長為572 bp,其中最長一處位于trnY和trnL1之間(間隔序列長度為225 bp);基因重疊有5處,共63 bp,nad4L和nad4之間重疊最長為40 bp;既無重疊又無間隔的區域有6處(表3)。
2.2" 分子標記
對腸舌狀絳蟲基因組的12 個PCGs 和2 個rRNA 基因的差異位點分析見表5。由表5可知,12 個PCGs中差異位點比例較高的3個基因分別為nad2(22.59%)、nad6(20.92%)、nad4(20.30%),nad4L基因最保守,差異位點比例僅為0.44%;2個rRNA 基因的差異位點比例均低于10%。
2.3" 腸舌狀絳蟲的系統發育關系
以長尾窄體吸蟲(Lyperosomum longicauda)為外群,通過ML和BI構建系統發育進化樹,兩種方法都生成具有一致分支拓撲的系統發育圖,因此僅顯示了后者(圖4)。系統發育樹結果顯示,在裂頭科絳蟲(Diphyllobothriidae)中,腸舌狀絳蟲(Ligula_intestinalis,NC_039445.1)和雙線絳蟲(Digramma)位于同一分支,本研究的分離蟲株(PP109086.1)再與該分支聚成一支,節點分支具有很高的可信度(BP = 100和PP = 1.00),并與雙槽頭屬絳蟲(Dibothriocephalus)形成姐妹群。該分支與復殖孔屬絳蟲(Diplogonoporus)聚成一支,然后與迭宮屬絳蟲(Spirometra)形成姐妹群。
3" 討" 論
線粒體基因組具有保守性高、母系遺傳、較快的進化速率等諸多優勢,使得在物種進化關系、物種鑒定以及揭示線粒體基因組和宿主適應關系等研究發揮了重要作用。此外,完整的線粒體基因組不僅可以揭示一般基因組特征,而且具有研究功能分子機制的潛力[21]。本研究測定了腸舌狀絳蟲線粒體基因組全序列,全長為13 655 bp,比分離自中國的青海湖裸鯉(NC_039445.1)和俄羅斯的拉多加環斑海豹(MW602519.1、MW602519.1)長(表1),但是本研究的腸舌狀絳蟲的nad3基因和rrnS基因序列長度更短(詳見OSID開放科學數據與內容中的附件1,掃首頁OSID碼可獲取,下同)。堿基組成呈現出Tgt;Agt;Ggt;C,A+T含量為67.6%,具有明顯的AT偏好性,AT偏斜度為-0.305,GC偏斜度為0.245,表明整體更偏好使用T和G堿基。對其它4條腸舌狀絳蟲的線粒體基因組進行分析也得出相同的結論(詳見附件1)。而在線粒體基因組PCGs的密碼子第3位AT含量最高,與其它絳蟲一致[11]。腸舌狀絳蟲的線粒體基因組缺少atp8基因,除了cox3基因使用GTG作為起始密碼子外,其它PCGs都使用ATG作為起始密碼子,這些特征均與其它扁形動物一致[22]。本研究中分離的腸舌狀絳蟲線粒體基因組中5 個PCGs使用TAA作為終止密碼子,5 個PCGs使用TAG,而cox3基因和nad3基因都沒有終止密碼子。在GenBank數據庫中其它4條腸舌狀絳蟲線粒體基因組(NC_039445.1,MW602519.1,MW602520.1,OR756289.1)的cox3基因則使用不完全密碼子T--,其中3條線粒體基因組的nad3基因以TAG作為終止密碼子,1條線粒體基因組的nad3基因的終止密碼子為T--,這與本研究不一致(詳見附件1)。
腸舌狀絳蟲無特異性宿主,大約有170多種魚可作為其第二中間宿主,以食魚鳥作為終末宿主[2]。本研究中用于比較的其中兩條腸舌狀絳蟲分離自拉多加環斑海豹,這種哺乳動物是腸舌狀絳蟲的偶然宿主[23]。由于腸舌狀絳蟲的寄生宿主跨度極大,因此,我們進一步將分離自拉多加環斑海豹的兩株腸舌狀絳蟲(MW602519.1、MW602520.1)的線粒體基因組與分離自青海湖裸鯉的腸舌狀絳蟲(NC_039445.1)進行比較,發現它們的線粒體基因組結構和每個基因的長度完全相同(詳見附件1)。環境的物理屏障和地理隔離可能會導致寄生蟲的遺傳差異[24],但是對于腸舌狀絳蟲卻不是這樣。由于GenBank數據庫中現有的腸舌狀絳蟲的全基因組序列只有5條,需要更多的數據來支撐以上結論。
對腸舌狀絳蟲線粒體基因組的密碼子偏好性進行分析,發現在62個編碼的密碼子中有23個密碼子的RSCU值大于1,其中密碼子UUA的RSCU值最高,而密碼子CGA的RSCU值則最低。一般認為如果密碼子沒有偏好,則該密碼子的RSCU值等于1。當密碼子的RSCU值大于1時,表明該密碼子具有較高的使用率,反之亦然[23]。因此,有23個密碼子是腸舌狀絳蟲的偏好密碼子。進一步分析發現,腸舌狀絳蟲和雙線絳蟲均以UUA作為最優密碼子,本文中選取的用于構建進化樹的蟲種(表2)均使用UUA作為優勢密碼子,并且RSCU值高的密碼子也幾乎以A或U結尾,這與PCGs的堿基呈AT偏向性的分析結果一致(表4)。對22個tRNA的二級結構進行分析發現,除trnS1和trnR外,其余20個tRNA均可形成典型的三葉草結構,這種現象也存在于裂頭科絳蟲、頭頰科絳蟲和帶形科絳蟲[11-12,25]。
合適的分子標記對于群體遺傳學研究至關重要,甚至可能在基因同源性分析中得出不同的結論[5]。張學勇等[5]基于18S rRNA、cox1和cytb基因這3個基因的聯合分析對舌狀絳蟲分離株進行分子進化發育研究,發現采用不同的分子標記基因可得出不同的結論,主要是由于核基因分子標記變異小,而線粒體基因標記變異較大。本研究對GenBank數據庫中現有的5條腸舌狀絳蟲的線粒體全基因組序列(包含本研究中上傳的序列)的12 個PCGs 和2 個rRNA 基因的差異位點進行分析,發現nad4L基因最保守,不適合用于物種同源性分析,而較為理想的分子標記包括nad2、nad6和nad4基因,可用于物種同源性分析。
本研究構建的線粒體全基因組12個PCGs的核苷酸序列的系統發育樹結果表明腸舌狀絳蟲與雙線絳蟲的親緣關系最接近。Li等[11]通過比較腸舌狀絳蟲與雙線絳蟲的線粒體全基因組序列結合系統發育樹,認為二者應歸屬于同一個屬。在更早的研究中,Li和Liao[6]通過比較腸舌狀絳蟲與雙線絳蟲的28S rRNA、cox1、ITS1和ND1基因,發現兩種絳蟲的4個基因都是高度保守的, 4個基因的低遺傳差異表明腸舌狀絳蟲與雙線絳蟲應該都歸屬于舌狀屬。本研究的結果與這兩篇文獻報道一致。除了雙線絳蟲,腸舌狀絳蟲與雙槽頭屬絳蟲形成姐妹群。通過串聯雙槽頭屬絳蟲和舌狀屬絳蟲18S rDNA+28S rDNA+rrnL+cox1的核苷酸序列,同樣證明雙槽頭屬絳蟲是舌狀屬絳蟲的姐妹群[26]。在裂頭科絳蟲中,復殖孔屬、舌狀屬、雙葉槽屬和迭宮屬聚成一大分支,這與前人的報道也一致[11,25]。腸舌狀絳蟲被認為是研究寄生蟲生態學和遺傳學的良好模型[27],因此,有必要對腸舌狀絳蟲作進一步的深入研究。
4" 結" 論
腸舌狀絳蟲的線粒體基因組全長為13 655 bp,其堿基組成具有明顯的AT偏向性,包括12個PCGs、22個tRNA基因、2個rRNA基因和2個非編碼控制區。nad2、nad6和nad4基因更適合作為腸舌狀絳蟲的分子標記。腸舌狀絳蟲與雙線絳蟲親緣關系最近。本研究為腸舌狀絳蟲的分類地位、系統發育分析和線粒體基因組研究奠定了堅實的基礎。
參考文獻(References):
[1]" LAGRUE C, PRESSWELL B, DUNCKLEY N, et al. The invasive cestode parasite ligula from salmonids and bullies on the South Island, New Zealand[J]. Parasitol Res, 2018, 117(1):151-156.
[2]" KAPUSTA A, BOGACKA-KAPUSTA E, CZARNECKI B. The significance of stone moroko, Pseudorasbora Parva (temminck and schlegel), in the small-sized fish assemblages in the littoral zone of the heated Lake Licheńskie[J]. Arch Pol Fish, 2008, 16(1):49-62.
[3]" GUTIRREZ J S, HOOLE D. Ligula intestinalis[J]. Trends Parasitol, 2022, 38(4):344-345.
[4]" 申" 夕, 饒天宇, 許" 薇, 等. 蕪湖間斷雙線絳蟲裂頭蚴的形態和分子特征及系統發育研究[J]. 齊齊哈爾醫學院學報, 2020, 41(11):1321-1325.
SHEN X, RAO T Y, XU W, et al. Characteristics of morphology and molecular and phylogenetic analysis of the Digramma interrupta plerocercoid in Wuhu[J]. Journal of Qiqihar Medical University, 2020, 41(11):1321-1325. (in Chinese)
[5]" 張學勇, 簡瑩娜, 李秀萍, 等. 青海湖裸鯉舌狀絳蟲裂頭蚴的分子鑒定及系統發育研究[J]. 水生生物學報, 2018, 42(1):33-38.
ZHANG X Y, JIAN Y N, LI X P, et al. Molecular identification and phylogenetic study of Ligula intestinalis pleroceroid in Gymnocypris przewalskii from the Qinghai Lake, China[J]. Acta Hydrobiologica Sinica, 2018, 42(1):33-38. (in Chinese)
[6]" LI J, LIAO X. The taxonomic status of Digramma (Pseudophyllidea: Ligulidae) inferred from DNA sequences[J]. J Parasitol, 2003, 89(4):792-799.
[7]" LOGAN F J, HORK A, TEFKA J, et al. The phylogeny of diphyllobothriid tapeworms (Cestoda:Pseudophyllidea) based on ITS-2 rDNA sequences[J]. Parasitol Res, 2004, 94(1):10-15.
[8]" JIMNEZ-AVALOS G, SOTO-OBANDO A, SOLIS M, et al. Assembly and phylogeographical analysis of novel Taenia solium mitochondrial genomes suggest stratification within the African-American genotype[J]. Parasit Vectors, 2023, 16(1):349.
[9]" ZHOU X, WANG Z, ZHU P C, et al. Eimeria zuernii (Eimeriidae: Coccidia):mitochondrial genome and genetic diversity in the Chinese yak[J]. Parasit Vectors, 2023, 16(1):312.
[10]" 伊平昌, 李國平, 顧冬花, 等. 青海大通北川河源區魚類舌狀絳蟲和雙線絳蟲的分子鑒定及系統發育關系研究[J]. 中國獸醫雜志, 2019, 55(11):17-21.
YI P C, LI G P, GU D H, et al. Molecular identification and phylogenetic research of Ligula intestinalis and Digramma interuota in fish from the Qinghai Datongbeiheyuanqu Nature Reserve, China[J]. Chinese Journal of Veterinary Medicine, 2019, 55(11):17-21. (in Chinese)
[11]" LI W X, FU P P, ZHANG D, et al. Comparative mitogenomics supports synonymy of the genera Ligula and Digramma (Cestoda:Diphyllobothriidae)[J]. Parasit Vectors, 2018, 11(1):324.
[12]" CAO Z Y, XI B W, LI S W, et al. Characterization of the complete mitochondrial genome of Nippotaenia mogurndae Yamaguti and Miyata, 1940 (Cestoda: Nippotaeniidae)[J]. J Helminthol, 2022, 96:e65.
[13]" BOHARD L, LALLEMAND S, BORNE R, et al. Complete mitochondrial exploration of Echinococcus multilocularis from French alveolar echinococcosis patients[J]. Int J Parasitol, 2023, 53(10):555-564.
[14]" 葉玉龍. 6種肉孢子蟲線粒體基因組組成和結構分析[D]. 昆明:云南大學, 2020.
YE Y L. Analysis of the composition and structure of mitochondrial genomes of 6 species of Sarcocystis[D]. Kunming:Yunnan University, 2020. (in Chinese)
[15]" XIANG C Y, GAO F L, JAKOVLIC′ I, et al. Using PhyloSuite for molecular phylogeny and tree-based analyses[J]. iMeta, 2023, 2(1):e87.
[16]" TALAVERA G, CASTRESANA J. Improvement of phylogenies after removing divergent and ambiguously aligned blocks from protein sequence alignments[J]. Syst Biol, 2007, 56(4):564-577.
[17]" KALYAANAMOORTHY S, MINH B Q, WONG T K F, et al. ModelFinder: fast model selection for accurate phylogenetic estimates[J]. Nat Methods, 2017, 14(6):587-589.
[18]" TRIFINOPOULOS J, NGUYEN L T, VON HAESELER A, et al. W-IQ-TREE: a fast online phylogenetic tool for maximum likelihood analysis[J]. Nucleic Acids Res, 2016, 44(W1):W232-W235.
[19]" RONQUIST F, TESLENKO M, VAN DER MARK P, et al. MrBayes 3. 2: efficient Bayesian phylogenetic inference and model choice across a large model space[J]. Syst Biol, 2012, 61(3):539-542.
[20]" LETUNIC I, BORK P. Interactive tree of life (iTOL) v5: an online tool for phylogenetic tree display and annotation[J]. Nucleic Acids Res, 2021, 49(W1):W293-W296.
[21]" KAARNIRANTA K, PAWLOWSKA E, SZCZEPANSKA J, et al. Role of mitochondrial DNA damage in ROS-mediated pathogenesis of age-related macular degeneration (AMD)[J]. Int J Mol Sci, 2019, 20(10):2374.
[22]" LE T H, BLAIR D, MCMANUS D P. Mitochondrial genomes of parasitic flatworms[J]. Trends Parasitol, 2002, 18(5):206-213.
[23]" YU T H, LI J S, YANG Y, et al. Codon usage patterns and adaptive evolution of marine unicellular cyanobacteria Synechococcus and Prochlorococcus[J]. Mol Phylogenet Evol, 2012, 62(1):206-213.
[24]" FRAIJA-FERNNDEZ N, WAESCHENBACH A, BRISCOE A G, et al. Evolutionary transitions in broad tapeworms (Cestoda: Diphyllobothriidea) revealed by mitogenome and nuclear ribosomal operon phylogenetics[J]. Mol Phylogenet Evol, 2021, 163:107262.
[25]" LI W X, ZHANG D, BOYCE K, et al. The complete mitochondrial DNA of three monozoic tapeworms in the Caryophyllidea: a mitogenomic perspective on the phylogeny of eucestodes[J]. Parasit Vectors, 2017, 10(1):314.
[26]" WAESCHENBACH A, BRABEC J, SCHOLZ T, et al. The catholic taste of broad tapeworms-multiple routes to human infection[J]. Int J Parasitol, 2017, 47(13):831-843.
[27]" HOOLE D, CARTER V, DUFOUR S. Ligula intestinalis (Cestoda: Pseudophyllidea): an ideal fish-metazoan parasite model?[J]. Parasitology, 2010, 137(3):425-438.
(編輯" 白永平)
