宏蛋白質組學:研究微生物群落的一種新策略
2010-01-12 09:06:18劉虎虎盧向陽
微生物學雜志 2010年5期
劉虎虎,田 云,盧向陽,方 俊
(1.湖南省農業生物工程研究所,湖南長沙 410128;2.湖南農業大學生物科學技術學院,湖南長沙 410128)
1 宏蛋白質組學的產生
微生物世界是分子多樣性最大的天然資源庫,由于地球上絕大部分微生物(約99%)是未培養微生物,即指那些利用分子生物學技術能夠檢測到,迄今所采用的微生物純培養分離、培養方法還未獲得純培養的微生物。因此,傳統的分離培養方法極大地限制了人們認識微生物世界的視野。由于未培養微生物在自然環境微生物群落中占有非常高的比例,無論是其物種類群,還是新陳代謝途徑、生理生化反應、產物等都存在著不同程度的差異性和豐富的多樣性,因而其中勢必蘊涵著巨大的可開發生物資源。因此,如何有效開發利用這些微生物資源已受到廣大科研人員的高度重視。隨著生命科學與生物技術的飛速發展,多種分子生物學技術先后應用于微生物,特別是未培養微生物的研究,如利用16S rRNA分析復雜生態環境中未知微生物的種類。1994年,澳大利亞科學家W ilkins和W illiams[1]提出蛋白質組的概念,指一個基因組所表達的所有蛋白質,或細胞、組織或機體在特定時空所表達的全部蛋白質。蛋白質組學是研究一種細胞乃至一種生物所表達的全部蛋白質的生理結構與代謝過程的科學,但是對于組成復雜的天然樣品或含有大量不可培養微生物的自然微生物區系,蛋白質組學研究還很不足。1998年,Handels man等[2]首次正式提出宏基因組的概念,最初用來定義土壤細菌混合基因組,研究對象是生境中全部微小生物遺傳物質的總和(the genomes of the total microbiota found in nature),主要包括環境樣品中的細菌和真菌。現在宏基因組是指特定環境中所有生物遺傳物質的總和,以生態環境中全部DNA作為研究對象[3],避開了微生物分離培養的問題,極大地擴展了微生物資源的利用空間。目前宏基因組主要指環境樣品中的細菌和真菌的基因組總和,也可指特定環境或共生體內所有生物遺傳物質的總和,是一種不依賴于人工培養的微生物基因組分析技術[3]。而宏基因組學是一種以環境樣品中的微生物群體基因組為研究對象,以功能基因篩選和測序分析為研究手段,以微生物多樣性、種群結構、進化關系、功能活性、相互協作關系及與環境之間的關系為研究目的的新的微生物研究方法。宏基因組學的出現大大促進了對復雜微生物多樣性及其遺傳物質的研究,它在一定程度上彌補了傳統研究方法所存在的局限性。宏基因組學的出現標志著基因組學分析從單一微生物擴展到復雜的微生物區系,但是它不能從基因水平上很好地解釋系統的動態性。2004年,Rodriguez-Valera[4]在宏基因組和蛋白質組基礎之上,進一步提出了宏蛋白質組的概念,它是指環境混合微生物群落中所有生物的蛋白質組總和。宏蛋白質組學則是針對某一特定地點的微生物群落所產生的全部蛋白質而進行大規模研究的方法[5]。宏蛋白質組學與宏基因組學在生物基因表達與功能水平上是相對應的,兩者都是針對微生物特定時期動態的生命活動而進行的研究。兩者對應關系[6]如圖1所示。
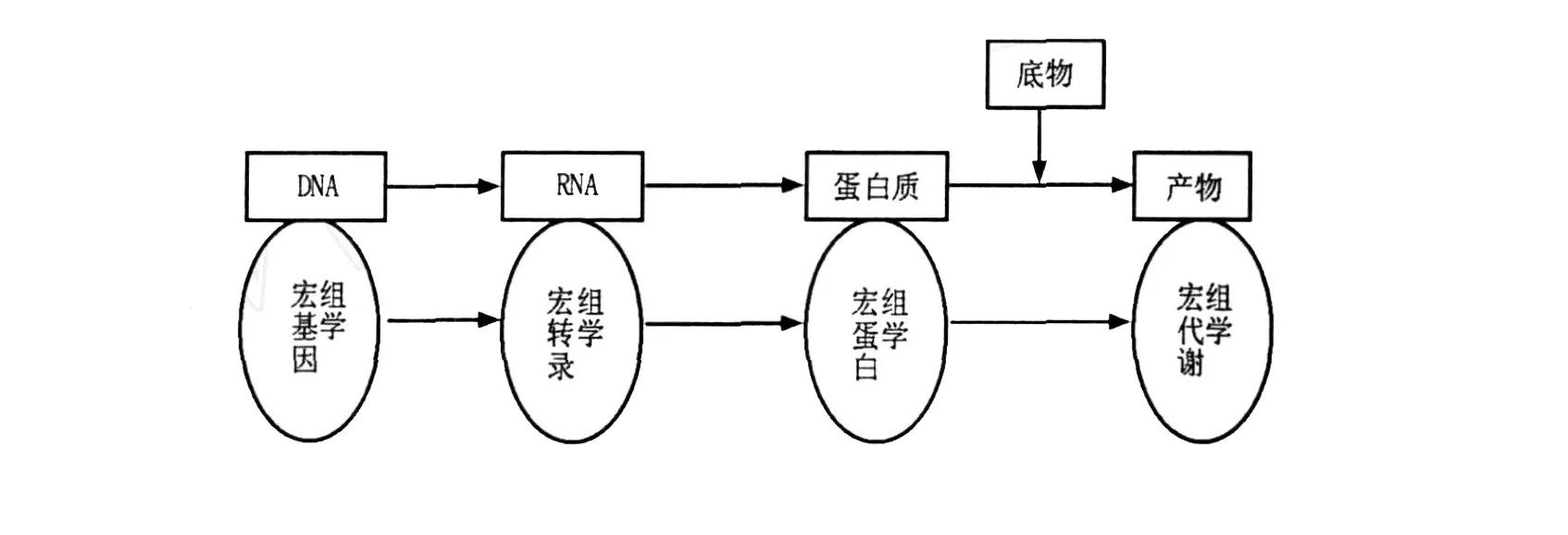
圖1 微生物群落基因功能表達與研究示意圖Fig.1 Schematic expression and research of gene in the ecology ofmicrobial communities
2 宏蛋白質組學研究策略及應用
2.1 宏蛋白質組學研究的基本技術路線及其策略
用于研究微生物群落的宏蛋白質組學與普通蛋白質組學在思路上基本相同。根據蛋白質理化性質的差異與不同的技術手段設計實驗路線。首先,對環境混合微生物群落中所有生物的蛋白質進行提取與純化。宏蛋白質組學的研究對象是特定環境中微生物所產生的所有蛋白質,因此,蛋白質的提取與純化是宏蛋白質組學研究的關鍵,但目前尚未存在一種通用的方法用于對全部蛋白質的提取。2009年,Abram等[7]采用聲波降解法與弗氏壓碎器破碎法提取總蛋白質,并應用宏蛋白質組技術研究在污水處理過程中微生物厭氧消化時蛋白質的表達情況,實驗結果表明,經聲波降解法所提取的蛋白質能夠增加雙向電泳分離蛋白質效果;其次,對所有蛋白質進行分離。目前蛋白質的分離一般采用雙向電泳與色譜技術;最后對蛋白質進行鑒定與分析。對于蛋白質的鑒定主要依賴于質譜技術。比較常用的質譜技術包括基質輔助激光解析電離-飛行時間質譜(Matrix-assisted laser desorption ionization time-of-flight mass spectrometry,MALD I-TOF-MS)、電噴霧質譜(Electrospray ionisation mass spectrometry,ESI-MS)與四極桿-飛行時間串聯質譜(Quadrupole t ime-of-flight mass spectrometry,Q-TOF-MS)等。將經過質譜技術所鑒定的蛋白質序列輸入數據庫進行檢索即可獲得目的蛋白的相關信息,但目前蛋白質數據庫中的蛋白質大多來源于已培養微生物,數據庫中包含的未培養微生物蛋白質的信息非常少,這是現在宏蛋白質組學研究面臨的一個重大問題[8]。宏蛋白質組學研究的基本技術路線如圖2所示[9]。W i lmes提到宏蛋白質組學的研究主要有兩個策略:一個是基于雙向電泳技術分析鑒定微生物蛋白質表達;另一個則是基于液相色譜技術分析鑒定微生物蛋白質表達[10]。
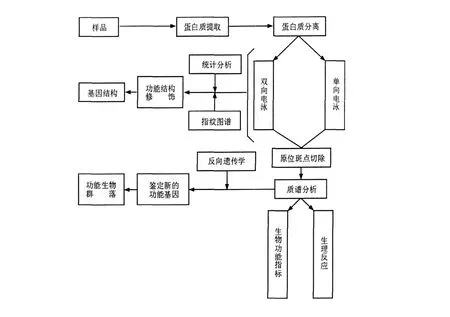
圖2 宏蛋白質組學研究基本技術路線示意圖Fig.2 Schematic approach of metaproteomics technique
2.2 宏蛋白質組學的應用
2.2.1 宏蛋白質組學在活性污泥微生物研究中的應用 Wilmes等[11]運用宏蛋白質組學技術研究活性污泥系統在強化生物除磷(Enhanced biological phosphorus removal,EBPR)過程中微生物蛋白質的表達情況。活性污泥在厭氧環境與好氧環境交替進行過程中可以增強生物除磷,在該過程中活性污泥微生物的蛋白質發揮著重要作用。經雙向電泳和質譜技術鑒定出3個差異蛋白質,進一步通過數據庫搜索和生物信息學分析推測這些蛋白質可能由β-變形細菌合成。W ilmes等[12]進一步運用雙向電泳與熒光原位雜交技術研究不同處理方法對活性污泥微生物總蛋白質表達的影響,經反應器處理的活性污泥定義為“EBPR”樣品,未經反應器處理的活性污泥定義為“nEBPR”樣品,反應器可使活性污泥中微生物在厭氧環境與好氧環境不同兩相中交替進行生物除磷。實驗結果證明經反應器處理的“EBPR”樣品分離得到630個蛋白質,反應前后存在表達差異的蛋白質占9.4%;而未經反應器處理的“nEBPR”樣品分離得到590個蛋白質,存在表達差異的蛋白質占14.7%。在此基礎之上,W ilmes等[13]運用雙向電泳與質譜技術鑒定活性污泥中微生物在生物除磷過程中發揮重要作用的蛋白質,共鑒定出與生物除磷過程相關的蛋白質46個,這些蛋白質可能分別參與糖原的合成與分解、三羧酸循環、脂肪酸的合成等生化反應。Park等[14]利用宏蛋白質組學技術系統研究活性污泥蛋白質在厭氧與好氧等不同環境中的表達情況,并運用液相色譜-串聯質譜法(Liquid chromatography-mass spectrography,LC-MS/MS)對差異蛋白質進行鑒定與分析,發現其中1個差異蛋白質為巴氏甲烷八疊球菌甲基化輔酶M還原酶(Methyl-coenzyme M reductase,MCMR)的亞基。
2.2.2 宏蛋白質組學在微生物起源研究中的應用 Kan等[15]利用雙向電泳和LC-MS/MS對切薩皮克海灣不同水域中微生物群落進行研究,發現該海灣不同水域所分離的微生物蛋白質的表達存在差異,經質譜鑒定表明這些差異蛋白質與已知數據庫的蛋白質不匹配,經進一步鑒定與生物信息學分析,其中3個蛋白質(CB1、CB3、CB6)可能與海洋微生物起源有關,其中CB3可能是NADH-輔酶Q氧化還原酶的亞基,而CB6與氨肽酶功能相類似。
2.2.3 宏蛋白質學在微生物脅迫應答中的應用Lacerda等[16]運用雙向電泳、MALD I-TOF-MS與肽端從頭測序技術研究重金屬鎘對細菌生物群落蛋白質表達的影響,結果發現重金屬鎘處理0.25 h與3 h時微生物的蛋白質表達發生顯著變化,一共鑒定出109個差異表達蛋白質,這些蛋白質包括ATP酶、氧化還原酶和轉運蛋白等,這是首次應用從頭測序技術在大規模范圍內研究未知微生物蛋白質的動態表達情況。
2.2.4 宏蛋白質組學在腸道微生物研究中的應用 Klaassene等[17]利用雙向電泳與質譜技術分析了一個家庭中嬰兒A和嬰兒B在斷奶之前不同時間排泄物中未培養微生物的生長情況,研究發現排泄物在不同時期所含微生物的蛋白質表達存在著一定的差異。研究以嬰兒A 41 d排泄物為樣品,應用MALD I-TOF-MS鑒定55個蛋白點并發現其與數據庫不匹配。對于嬰兒A不同時期(8、24、41 d)相同的4個蛋白質位點(1、2、7、11)進行鑒定分析發現其與已報道蛋白質存在差異。與此同時,研究發現1個含有肽端序列的蛋白質與雙歧桿菌的轉移酶相類似。Verberkmoes等[18]利用宏蛋白質組學技術研究幼兒雙胞胎各自排泄物中蛋白質的表達情況,并以此推測人體腸道微生物的代謝情況,結果發現宏蛋白質組學相對于宏基因組學呈非正態分布。此外,研究發現樣品中部分蛋白質屬于同源蛋白,它們可能參與人體免疫和微生物代謝活動。實驗之所以選擇人體排泄物作為樣品,是因為排泄物具有非侵入性,而且樣品中部分蛋白質經鑒定分析與人體內蛋白質相同,這些蛋白質具有一定的代表性。
2.2.5 宏蛋白質組學在口腔微生物研究中的應用 J.D.Rudney等[19]應用三維肽段分離技術與串聯質譜技術對分別取自口腔癌患者與正常人群唾液樣品微生物進行宏蛋白質組分析。實驗主要以口腔癌患者唾液為研究對象,并根據蛋白質理化性質差異分離鑒定出7 000個多肽,其中357個多肽與微生物起源相關。研究通過系統進化方法分析這些多肽發現其主要屬細菌類群。在種分類學劃分水平,僅鑒定出11%多肽;在門分類學劃分水平,29%多肽屬厚壁門,其中大部分多肽屬鏈球菌屬;20%多肽屬變形菌門;4%多肽屬放線菌門;3%多肽屬擬桿菌門;1%多肽屬螺旋體門等。研究經通路分析技術發現同源蛋白質簇(Clusters of Orthologous Groups,COGs)中37%多肽屬J組,參與蛋白翻譯;19%多肽屬G組,參與糖酵解過程;8.2%多肽屬E組,參與氨基酸代謝;7.9%多肽屬C組,與產能有關。此外,研究還發現包括蚜蟲內生共生體、極端微生物與古細菌在內的部分新物種。
2.2.6 宏蛋白質組學在膜蛋白研究中的應用RobertM Morris等[20]應用串聯質譜技術對南大西洋10個不同區域樣品表層水膜蛋白進行研究分析。通過構建16S rRNA文庫并對849個16S rRNA序列比對分析發現其中184個序列屬放線菌;31個序列屬細菌浮游生物;90個序列屬綠球藻;177個序列屬SAR11。它們的分布情況反映出不同海域富含不同的營養物質。作者對來自10個樣品共5 389個肽段進行鑒定并對939個特殊肽段作進一步分析,發現這些肽段大多屬TBDTs(TonB-dependent transporters)、孔蛋白與ABC轉運蛋白。這些蛋白質主要參與物質運輸過程,其中TBDTs通過質子動力勢轉運營養物質。研究采用量多元尺度法與串聯質譜技術聯合分析發現不同蛋白在沿海水域與公海水域含量存在差異。同時,研究發現不同水域樣品中均含有視紫紅質并推測光營養細菌浮游生物可以利用光能參與不同形式的生命活動。此外,在南大西洋上層流發現古細菌氨單加氧酶與病毒蛋白,據此推測古細菌作為一類重要的硝化細菌在富含營養的表層水中起著重要的作用。
3 存在問題與展望
宏基因組學為微生物資源的開發利用提供了重要的信息,同時也為未培養微生物的研究提供了新的技術手段,但目前尚無較好方法用于研究微生物與環境之間的相互作用。宏蛋白質組學概念的提出為解決這一難題提供了可能,它以微生物群落所產生的所有蛋白質為研究對象,應用宏蛋白質組技術對其進行鑒定分析并研究微生物在復雜環境中生命活動的動態變化。宏蛋白質組學的研究工作目前尚處于初級階段,仍存在許多問題有待解決。一方面來自微生物內因,地球上約99%微生物屬未培養微生物,而且幾乎所有的微生物在其生命周期中都會遇到各種不同的環境。這些微生物需要適當調整其所表達的蛋白質,從而維護細胞內部的代謝平衡。大多數微生物在不同的生長環境下僅表達它們整個蛋白質組中的一部分,較難獲得環境樣品中微生物所表達的高宏蛋白質組覆蓋率;另一方面來自研究技術的局限性。比如雙向凝膠電泳技術不能夠檢測低豐度蛋白或具有極端物理化學屬性的蛋白,值得一提的是多維蛋白鑒定技術與GeLC-MS/MS方法在上述方面已取得一些進步。此外,用來將肽段與特異性蛋白進行匹配的生物信息學方法可能會產生假陽性鑒定結果,從而影響對于蛋白質的分析鑒定。因此,解決這些問題成為推動宏蛋白質組學技術發展的一個主要力量。隨著科學技術的不斷發展,宏蛋白質組學將在微生物群落研究中發揮越來越重要的作用。
[1] Graves PR,Haystead TA.Molecular biologist’s guide to proteomics[J].MicrobiolMolBiol Rev,2002,66(1):39-63.
[2] Handels man J,Rondon MR,Brady SF,et al.Molecular biological access to the chemistry of unknown soil microbes:a new frontier for natural products[J].Chem Biol,1998,5:245-249.
[3] Rondon MR,August PR,Bettermann AD,et al.Cloning the soilmetagenome:a strategy for accessing the genetic and functional diversity of uncultured microorganis ms[J].Appl Environ Microbiol,2000,66:2541-2547.
[4] Rodríguez-Valera F.Environmental genomics,the big picture?[J].FEMS Microbiol Lett,2004,16:231(2):153-158.
[5] Wilmes P,Bond PL.The application of two-dimensional polyacrylamide gel electrophoresis and downstream analyses to a mixed community of prokaryotic microorganisms[J].Environ Microbiol,2004,6:911-920.
[6] Maron PA,Ranjard L,Mougel C,et al.Metaproteomics:a new approach for studying functional microbial ecology[J].Microb Ecol,2007,53:486-493.
[7] Abram F,Gunnigle E,O’Flaherty V.Optimisation of protein extraction and 2-DE for metaproteomics of microbial communities from anaerobic wastewater treatment biofilms[J].Electrophoresis,2009,30:4149-4151.
[8] 郝純,劉慶華,楊俊仕,等.宏蛋白質組學:探索環境微生態系統的功能[J].應用與環境生物學報,2008,14:270-275.
[9] Maron PA,Ranjard L,Mougel C,et al.Metaproteomics:a new approach for studying functional microbial ecology[J].Microb Ecol,2007,53:486-493.
[10] Wilmes P,Bond PL.Metaproteomics:studying functional gene expression in microbial ecosystems[J].Trends Microbiol,2006,14:92-97.
[11] Wilmes P,Bond PL.The application of two-dimensional polyacrylamide gel electrophoresis and downstream analyses to a mixed community of prokaryotic microorganis ms[J].Environ Microbiol,2004,6:911-920.
[12] Wilmes P,Bond PL.Towards exposure of elusive metabolic mixed-culture processes:the application of metaproteomic analyses to activated sludge[J].Water Sci Technol,2006,54:217-226.
[13] Wilmes P,Wexler M,Bond PL.Metaproteomics provides functional insight into activated sludge wastewater treatment[J].PLoSOne,2008,3:1-11.
[14] Park C,Helm RF.Application of metaproteomic analysis for studying extracellular polymeric substances(EPS)in activated sludge flocs and their fate in sludge digestion[J].Water Sci Technol,2008,57:2009-2015.
[15] Kan J,Hanson TE,Ginter JM,et al.Metaproteomic analysis of Chesapeake Bay microbial communities[J].Saline Systems,2005,1:7.
[16] Lacerda CM,Choe LH,Reardon KF.Metaproteomic analysis of a bacterial community response to cadmium exposure[J].J Proteome Res,2007,6:1145-1152.
[17] Klaassens ES,de VosWM,Vaughan EE.Metaproteomics approach to study the functionality of the microbiota in the human infant gastrointestinal tract[J].Appl Environ Microbiol,2007,73:1388-1392.
[18] Verberkmoes NC,Russell AL,Shah M,et al.Shotgunmetaproteomics of the human distal gut microbiota[J].IS ME J,2009,3:179-189.
[19] Rudney JD,Xie H,Rhodus NL,et al.A metaproteomic analysis of the human salivary microbiota by three-dimensional peptide fractionation and tandem mass spectrometry[J].Molecular Oral Microbiology,2010,25:38-49.
[20] Morris RM,Nunn BL,Frazar C,et al.Comparative metaproteomics reveals ocean-scale shifts in microbial nutrient utilization and energy transduction[J].The IS ME Journal,2010,4:673-685.
猜你喜歡
體育科技文獻通報(2022年3期)2022-05-23 13:46:54
中老年保健(2021年12期)2021-08-24 03:30:40
天津外國語大學學報(2021年3期)2021-08-13 08:32:18
遼金歷史與考古(2021年0期)2021-07-29 01:06:54
中國傳媒大學學報(自然科學版)(2021年1期)2021-06-09 08:43:00
中國生殖健康(2020年6期)2020-02-01 06:28:50
科技傳播(2019年22期)2020-01-14 03:06:54
新世紀智能(英語備考)(2019年12期)2020-01-13 06:07:18
民用飛機設計與研究(2019年4期)2019-05-21 07:21:24
中國生殖健康(2019年11期)2019-01-07 01:28:02
