飲用水源水中丁基黃原酸鹽測定關鍵問題探析
2010-09-15 16:09:44楊文武
環境影響評價 2010年2期
關鍵詞:標準
張 鈞,楊文武
飲用水源水中丁基黃原酸鹽測定關鍵問題探析
張 鈞,楊文武
(泰州市環境監測中心站,江蘇泰州225300)
應用GB/T5750.8-2006《銅試劑亞銅分光光度法》對飲用水源水中丁基黃原酸鹽的測定進行了系統的探析。探討了運用該方法測定飲用水源水中丁基黃原酸鹽進行的一些技術問題。在標準分析方法的基礎上,選用最新生產的合格的化學試劑,適當改進相關操作步驟,利用無水硫酸鈉粉末干燥有機相層并破除乳化現象,能夠確保水樣丁基黃原酸鹽測定的精密度和準確度。
丁基黃原酸鹽;可見光分光光度法;飲用水源水
丁基黃原酸鹽,俗稱“丁基黃藥”[1],是一種重要的金屬硫化礦捕集藥劑、橡膠硫化促進劑,廣泛應用于各種重金屬硫化礦(如PbS、ZnS、NiS、CuS等)和部分貴金屬硫化礦(如Au2S3、Ag2S等)的浮選捕收。黃藥因呈黃色故而得名。丁基黃原酸鹽在常溫下為淺黃色至黃色粉狀或棒粒狀固體,有毒,易燃,易吸潮,易溶于水、丙酮和部分醇中,性質不穩定,在酸性介質中加速分解。丁基黃原酸鹽對人體和畜禽的危害[2]主要表現在傷及神經系統和肝臟器官,對造血系統也有不良影響。在金屬硫化礦的浮選捕集過程中,丁基黃原酸鹽大部分留在礦石表面,只有很少部分隨廢水排入地表水,污染飲用水源和環境[3]。因而丁基黃原酸鹽已被列為集中式生活飲用水地表水源地特定監測項目之一。
對丁基黃原酸鹽的測定,目前大多采用銅試劑亞銅分光光度法[4]、萃取比色法、離子選擇電極法等。萃取比色法[5]過程比較繁瑣,條件苛刻,使用有毒有害的有機溶劑,危害操作人員身心健康,并對環境造成二次污染,不利于實際工作的開展;離子選擇性電極法[5]引起干擾的成分復雜,受待測水樣的p H、水溫、電極的響應時間等影響,水樣中較高濃度的鈣、鋁、銅和鐵離子等也均有干擾,特別是具有良好選擇性的電極較少。銅試劑亞銅分光光度法雖然靈敏度較低、重現性差,但因其具備投資費用少、便于推廣普及、適合大批量水樣分析和不需使用濃酸、濃堿、有機溶劑等有毒有害試劑等特點,而成為首選測定方法。本文主要以備受關注的飲用水源水為例,應用GB/T5750.8-2006《銅試劑亞銅分光光度法》進行飲用水源水中丁基黃原酸鹽測定的方法原理、儀器與試劑和一些關鍵操作技術問題的探討,同時給出了丁基黃原酸鹽測定過程中常見操作技術問題的解決方法。
1 方法原理
該方法的原理[4]是在p H=5.2的鹽酸羥胺還原體系中,將銅離子還原成亞銅離子。水樣中的丁基黃原酸鹽與亞銅離子生成黃原酸亞銅后,被環己烷萃取。黃原酸亞銅再與銅試劑作用,生成橙黃色的銅試劑亞銅,其色度與丁基黃原酸鹽含量成線性關系,符合朗伯-比爾定律,在波長436nm處有最大吸收,利用可見光分光光度計對水樣進行吸光度定量,從而實現丁基黃原酸鹽的測定。各階段化學反應方程式如下:
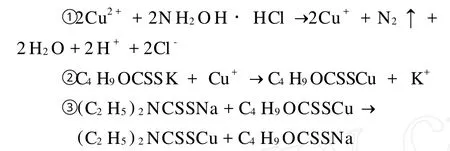
銅試劑亞銅(橙黃色)
2 儀器與試劑
2.1 儀器設備
p H計;抽濾裝置;10×30(內徑mm×長度cm)普通玻璃層析柱;樣品運輸貯藏箱(箱內溫度保持0~4℃,以冰袋制冷);分液漏斗振蕩器;其他所需儀器設備均同參考文獻[4]。
2.2 材料試劑
廣泛p H試紙:p H=1~14;純水:新制備的去離子水或蒸餾水;無水硫酸鈉(Na2SO4):在300℃的馬弗爐中烘烤2小時,轉移至干燥器中冷卻至室溫,裝入磨口玻璃瓶中,干燥器中保存,分析純;丁基黃原酸鹽:丁基黃原酸鉀,C4H9OCSSK,>95.0% (純度),日本東京化成工業株式會社生產,分析純;其他所需材料試劑及試劑配制方法均同參考文獻[4]。
3 關鍵操作技術探析
3.1 儀器設備的準備
測定中需使用的玻璃器皿(包括采樣瓶)要求內壁光潔無磨損,防止丁基黃原酸鹽被吸附。所有玻璃器皿應徹底洗凈,先用洗滌劑浸泡清洗,然后用(1 +9)鹽酸溶液或(1+35)硫酸溶液浸泡15 min,最后依次用自來水、純水沖洗干凈,這樣可以減少空白實驗的吸光度。玻璃器皿干燥后方能使用。天平、容量瓶、移液管、刻度吸管等計量儀器必須按有關規定檢定合格后方可使用。
3.2 水樣的采集和保存
水樣中丁基黃原酸鹽含量高低不等,且其易分解,在p H高于10或低于9時,分解加速,在酸性介質中則迅速分解生成CS2和相應的醇,而生成的醇可自動催化分解反應[6]。丁基黃原酸鹽水樣的采集與保存方法對分析結果有重大影響,具體可采用以下方法:把水樣采集在經充分洗凈并干燥的1000 mL棕色玻璃瓶中,立即放入冰箱中或樣品運輸貯藏箱中(箱內溫度控制在0~4℃)冷藏保存,24h內測定;若采樣后不能及時進行測定,水樣需放置較長時間,可用p H計或廣泛p H試紙測試其p H值,向水樣中滴加20 g/L(m/v)氫氧化鈉溶液或1%(v/ v)鹽酸溶液調節水樣p H至9~10(用廣泛p H試紙測試),立即放入冰箱中或樣品運輸貯藏箱中冷藏保存,1周內測定。
為保證所加保存劑的質量及避免水樣在采集運輸過程中受到玷污,要求采樣人員在現場以純水代替水樣作全程序空白試驗,全程序空白試驗樣品的測定值必須低于銅試劑亞銅分光光度法(GB/ T5750.8-2006)方法規定的檢出限(0.002 mg/L),否則應查找原因。
3.3 試劑的選擇要求
化學試劑(包括溶劑)在水質檢測分析中的地位極其重要。從取樣到樣品處理、樣品測定都離不開化學試劑,一旦使用的化學試劑質量、純度等達不到分析實驗要求,就會對測定結果造成顯著影響。在丁基黃原酸鹽的測定分析中應嚴格使用 GB/ T5750.8-2006《銅試劑亞銅分光光度法》所列出的各種最新生產的合格的試劑(分析純以上),筆者推薦使用國藥集團上海化學試劑有限公司生產的各類試劑或純度較高的進口分裝試劑,對本項目影響較大的試劑主要是鹽酸羥胺和丁基黃原酸鹽標準物質。
鹽酸羥胺試劑的質量對本項目至關重要,它直接影響到測定的成敗及結果的準確性。本測定中要求所用鹽酸羥胺試劑是最新生產的、合格的且在有效期范圍之內的。合格的鹽酸羥胺試劑應該為白色細結晶,但由于試劑質量存在差異,有些廠家、批號的試劑達不到這個要求,致使實驗失敗或靈敏度降低。因此,筆者推薦使用國藥集團上海化學試劑有限公司生產的分析純鹽酸羥胺。由于鹽酸羥胺試劑在空氣中易吸濕潮解而失效,使用完畢后應貯于干燥器中或于冰箱中2~5℃密封冷藏避光保存。
丁基黃原酸鹽標準物質的質量對本項目也非常重要,它直接影響到本項目標準溶液的配制與標準曲線的繪制。本測定中,筆者推薦使用日本東京化成工業株式會社出品的、最新生產的且在有效期范圍之內的丁基黃原酸鉀(簡稱PBX,分析純)。考慮到丁基黃原酸鉀試劑放置于空氣中易吸潮分解而失效,使用完畢后應立即于冰箱中2~5℃密封冷藏避光保存,丁基黃原酸鹽標準溶液宜現用現配。
3.4 水樣預處理方法
1)若水樣無色、透明、澄清,可直接量取適量以供分析;若水樣渾濁或含有不溶性物質,可采用以下兩種方法去除干擾:①自然沉降法,將水樣靜置沉降30分鐘后量取適量上清液供作分析;②抽濾處理法,將水樣經0.45μm濾膜抽濾處理,去除不溶性物質干擾,量取適量過濾后水樣供作分析,經抽濾預處理水樣須同時以純水經相同步驟做空白試驗。
2)別用p H計或廣泛p H試紙測試各水樣的p H值,用4 g/L(m/v)氫氧化鈉溶液或0.8%(v/v)鹽酸溶液調節水樣的p H值至5~6之間。
3)若水樣中 S2-質量濃度大于等于0.1μg/L將對測定產生負干擾[4],則需進行氯化處理,使水樣中游離氯為0.5 mg/L,即可消除S2-的干擾,經氯化處理的水樣,需同時以純水代替水樣做試劑空白。
4)賀心然[5]等通過大量實驗證實,在6.00 mg/ L的丁基黃原酸鹽標準溶液中分別加入20倍量(即120.00 mg/L,下同)的銅離子、30倍量的錳離子、100倍量的鋅離子、100倍量的鐵離子,在此反應體系中對丁基黃原酸鹽的測定無影響。
3.5 測定中的注意事項
3.5.1 溶解時間的控制
標準方法上指出[4]添加了鹽酸羥胺的標準管和水樣管,應于混勻后靜置30 min,以確保鹽酸羥胺固體試劑的充分溶解。筆者通過大量的分析實踐發現,由于鹽酸羥胺屬強還原劑,其長時間露置于空氣中易氧化分解而失效,因此,筆者建議鹽酸羥胺的溶解靜置時間不宜太長,放置3~5 min,待分液漏斗中氣泡消失即可。
3.5.2 振搖萃取方式的選擇
考慮到振蕩萃取效果的一致性與完全性,測定中,將待振蕩萃取的水樣和標準系列經預先振搖放氣兩次后,統一放置于分液漏斗振蕩器上,設置振蕩頻率為300次/分鐘,機械振搖4 min后,靜置分層。3.5.3 靜置分層時間的確定
靜置的目的是使不穩定的乳濁液分層。考慮到測定中需使用不穩定試劑及有不穩定產物生成,對于靜置分層時間的選擇,筆者通過大量的分析實踐認為,在將分液漏斗經過4分鐘劇烈機械振搖后,靜置至液體能夠分為清晰的上下兩層(有機相層與水相層),下層水相層清澈透明,且分液漏斗中氣泡消失即可,一般情況須靜置10 min,成分復雜水樣須靜置較長時間(30 min左右),以不超過 30 min為宜。
3.5.4 p H5.2純水的替換
銅試劑亞銅分光光度法(GB/T5750.8-2006)第三步中指出,在分去分液漏斗中水層后,向各分液漏斗中分別加入10 mL p H5.2的純水洗滌分液漏斗,振搖30 s,靜置分層。考慮到純水精密p H值定位工作的繁雜性,筆者通過大量的分析實踐認為, p H5.2的純水可以直接用p H5.2的乙酸-乙酸鈉緩沖溶液代替。
3.5.5 銅試劑加入量的量化
考慮到標準分析方法上規定加入少量,筆者通過大量的分析實踐認為,銅試劑(二乙基二硫代氨基甲酸鈉,簡稱DDTC)的加入量以50~60 mg為宜,與加入的有機相層后充分振蕩混勻后,至出現剩余少量DDTC未溶解即可。
3.5.6 萃取后乳化現象的消除
由于水樣組成復雜,有些水樣在加入有機溶劑進行振蕩萃取,靜置分層后,上層有機相層出現大量泡沫和小氣泡,下層水層也出現渾濁,即出現了乳化現象,形成乳濁液而難以分離。對此,標準分析方法中雖采用脫脂棉吸水以達到干燥有機相層的目的,但其未能有效消除萃取后有機相層中的乳化現象。筆者通過大量的分析實踐認為,由于無水硫酸鈉粉末極易吸水并能與之充分結合形成致密團塊,且無水硫酸鈉粉末呈中性,其不與有機相層中組分發生反應,因此,可以充分利用無水硫酸鈉粉末的這些特性以達到干燥有機相層、破除乳化的目的,具體做法如下:將經p H5.2的乙酸-乙酸鈉緩沖溶液三次充分振蕩洗滌的有機相層(徹底分去水層)自分液漏斗頸部放入到預先填充有3~5 cm高無水硫酸鈉粉末的玻璃層析柱中,使經無水硫酸鈉粉末充分吸水干燥后的有機相層放入到預先填充有50~60 mgDDTC和1滴純水的10 mL具塞比色管中,若有機相層在玻璃層析柱中出現滯流現象,可用洗耳球自玻璃層析柱上口吹入空氣,使有機相層在大氣壓的作用下緩緩放入到上述10 mL具塞玻璃磨口比色管中,充分振蕩比色管(此時應剩余少量DDTC未溶解),混勻,靜置顯色15 min。
經無水硫酸鈉粉末干燥破乳的水樣須同時以純水代替水樣做試劑空白。
3.5.7 吸光度測定時的注意事項
1)本項目中,水樣和標準系列吸光度的測定采用可見光分光光度計。使用中應保持光度計樣品池干燥,光度計所配備的干燥劑應定期更換。若干燥劑失效將導致:①數顯不穩,無法調“0”點或“100%”點(電路或光電管受潮);②反射鏡發霉或沾污,影響光效率,雜散光增加。鑒于上述原因,分光光度計的放置地點應遠離水池等濕度大的地方,干燥劑應定期更換或烘烤。
2)吸光度測定中,應保證比色皿不傾斜放置。稍許傾斜,就會使參比樣品與待測樣品的吸收光程長度不一致,還可能使入射光不能全部通過樣品池,導致測試比準確度不符合要求。
3)吸光度測定中,應保持比色皿透光面清潔,防止沾污的有色物質影響測定;測定標準系列時,應從低濃度測至高濃度。
4)吸光度測定中,應注意不可將10 mL具塞玻璃磨口比色管中的水層放入到比色皿中,因水會使分光光度計讀數值不穩,干擾測定。
3.5.8 改進法標準曲線的繪制
依據《銅試劑亞銅分光光度法 GB/T5750.8-2006》和本文提出的幾點改進步驟,采用最小二乘法可以得出如下丁基黃原酸鹽標準系列的線性回歸方程,丁基黃原酸鹽標準系列見表1。
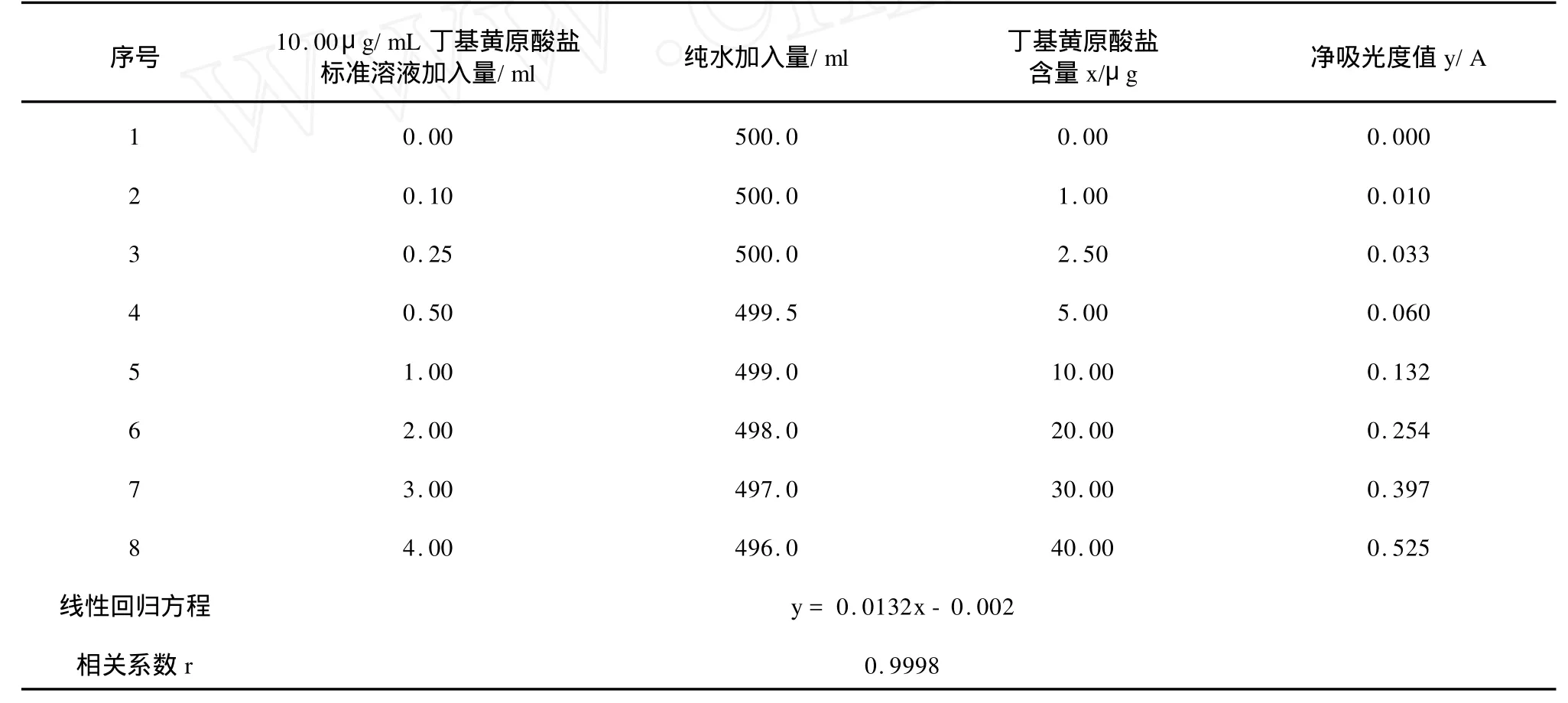
表1 丁基黃原酸鹽標準系列
3.5.9 改進法檢出限的測定
檢出限的測定目的是確定改進法測定的檢出限是否低于方法規定的檢出限;了解實驗用水、試劑、儀器、實驗室環境條件是否符合質量控制要求;了解分析人員的操作水平。
用純水代替水樣,與改進法標準曲線的繪制同步,每天測定2個空白平行樣,連測5天,計算10次空白平行樣吸光度對應的空白試驗濃度值的批內標準偏差Swb為0.00022 mg/L。5d空白平行測定值見下表2。
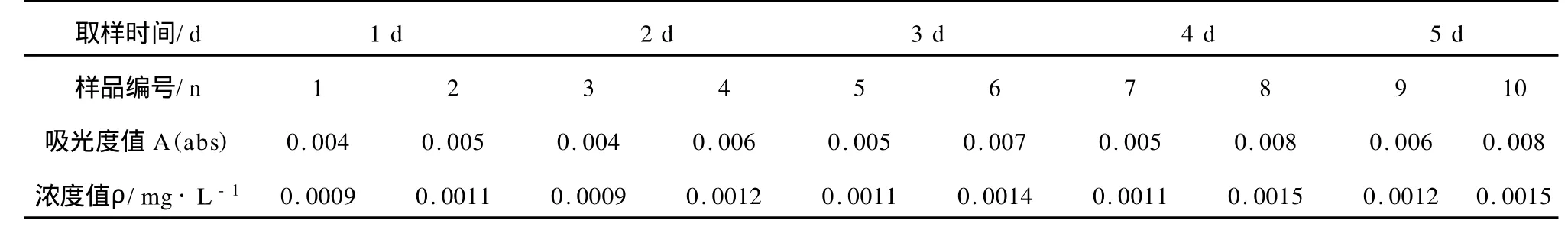
表2 5 d空白平行測定值
檢出限按下列公式計算:
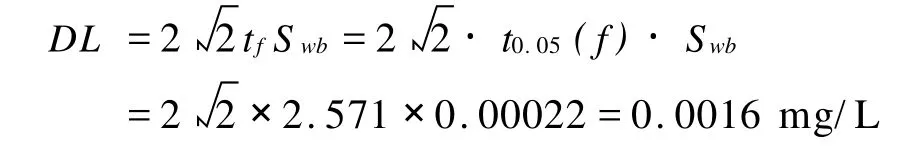
上式中:DL-方法的檢出限;t0.05(f)-單側顯著性水平為5%,批內自由度 f=m(n-1)時t分布臨界值(查 t分布表,當 f=5時,t0.05(f)= 2.571),m為重復測定次數,n為平行測定次數;Swb-10次空白試驗所測得濃度值的批內標準偏差;
改進法測定的檢出限(0.0016 mg/L)略低于原方法規定的檢出限(0.002 mg/L),可取為0.002 mg/L。
3.5.10 改進法精密度和準確度
利用已配制的100.0 mg/L的丁基黃原酸鹽標準儲備溶液經500倍和200倍兩個不同比例準確稀釋后,制得0.20 mg/L、0.50 mg/L兩種濃度的丁基黃原酸鹽標準溶液。采用上述丁基黃原酸鹽標準系列的測定程序對兩種濃度的丁基黃原酸鹽標準溶液進行6次平行測定,精密度試驗結果見表3。由表3可以看出,兩種不同濃度的丁基黃原酸鹽標準溶液6次測定的相對標準偏差均在3%以下,說明該方法具有較高的精密度。
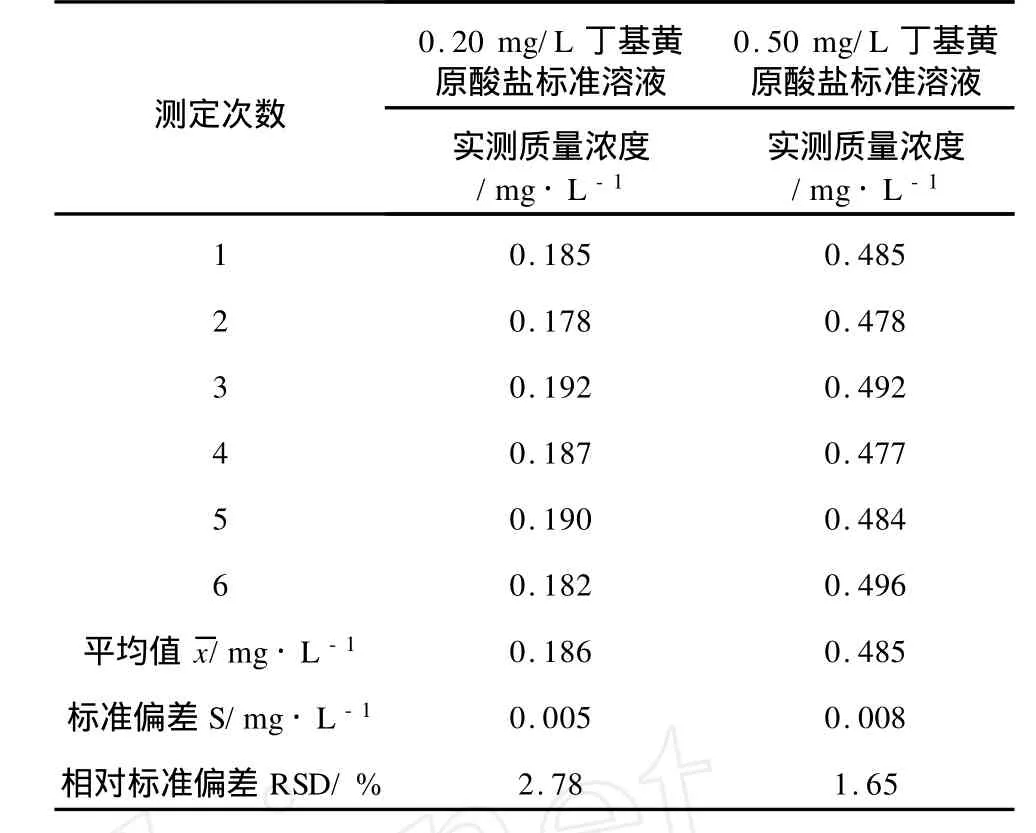
表3 精密度試驗結果
采用上述改進法丁基黃原酸鹽標準曲線對某地表飲用水源水樣1#、2#進行加標回收試驗,加標回收試驗結果列于表4。由表4可知,本方法的加標回收率為97.0%~98.5%,說明本方法具有較好的準確度。

表4 加標回收試驗結果
4 結語
本文結合自己的工作經驗和前人的研究成果,系統探討了應用國標分析方法—《銅試劑亞銅分光光度法 GB/T5750.8-2006》測定飲用水源水中丁基黃原酸鹽的方法原理、儀器試劑和一些關鍵操作技術問題。在標準分析方法的基礎上,應用改進法對丁基黃原酸鹽標準系列進行了標準曲線的繪制,對空白試驗中檢出限進行了測定,結果表明:改進法丁基黃原酸鹽標準曲線靈敏度高、線性相關性強(線性相關系數r=0.9998),改進法測定的檢出限(DL =0.0016 mg/L)略低于原方法規定的檢出限(DL =0.002 mg/L),符合檢出限測定質量控制要求。應用改進法對兩種不同濃度的丁基黃原酸鹽標準溶液進行6次平行測定,各標準溶液測定濃度的相對標準偏差均在3%以下;應用改進法對某地表飲用水源水樣1#、2#進行加標回收試驗,各水樣的加標回收率均在97.0%~99.0%之間,說明改進法具有較高的精密度和較好的準確度,符合分析質量控制的要求。
[1] 化工詞典.丁基黃藥[EB/OL].化工引擎,http://www.hellochem.com/xz/xz3/27646dimbg.htm.
[2] 環保技術聯合論壇.含黃藥廢水的處理方法和工藝[EB/OL].環保技術聯合網,http://bbs.epteks.com/topic.aspx?topicid =1719.
[3] 李廷才,鄒本崇.污水中丁基黃原酸鹽提取方法的探討-pH對測定的影響[J].數理醫藥學雜志,2000,13(04):351.
[4] 中華人民共和國衛生部,中國國家標準化管理委員會.中華人民共和國標準 生活飲用水標準檢驗方法有機物指標 GB/ T5750.8-2006丁基黃原酸銅試劑亞銅分光光度法[S].北京:中國標準出版社,2007.
[5] 賀心然,曹 雷,展衛紅,等.紫外分光光度法測定水中丁基黃原酸[J].環境污染與防治,2007,29(07):552-554.
[6] 陳景文,曹淑紅.滴定法測定堿金屬黃原酸鹽[J].理化檢驗-化學分冊,2004,40(06):355-357.
Determination of Butyl Xanthate in Drinking Source Water
(Taizhou Environmental Monitoring Center,Taizhou 225300,China)
ZHANGJun,YANG Wen-wu
Combined with the work practice of environmental monitoring,the application of the national standard classical analytical method-cupron-cuproine spectrophotometric method(GB/T5750.8-2006)to the determination of butyl xanthate in drinking source water was explorated systematically.On the base of standard method,chosing the latest produced and qualified chemical reagents,making appropriate improvements to the interrelated operating procedures,drying the organic medium phase and breaking through the emulsification in organic medium phase by the usage of decahedrons sodium sulfate powder,which can guarantee the accuracy and precision in the determination of butyl xanthate in water samples.
butyl xanthate;visible spectrophotometric method;drinking source water
X832
A
1674-2842(2010)02-0024-05
2009-11-17
張 鈞(1979-),男,漢族,江蘇姜堰人,碩士,助理工程師,從事環境監測工作。E-mail:thjzhangjun@163.com
猜你喜歡
城市道橋與防洪(2022年4期)2022-07-01 06:04:12
當代陜西(2019年8期)2019-05-09 02:22:48
上海建材(2019年1期)2019-04-25 06:30:48
動漫星空(興趣百科)(2019年3期)2019-03-07 07:23:10
家庭影院技術(2018年4期)2018-05-09 07:07:52
專用汽車(2016年4期)2016-03-01 04:13:43
質量與標準化(2015年9期)2015-12-31 11:41:40
中國質量與標準導報(2014年4期)2014-03-11 19:54:25
中國質量與標準導報(2014年10期)2014-02-28 22:25:47
中國質量與標準導報(2014年7期)2014-02-28 22:24:39
