
尿素在ZnO()表面的吸附
2010-12-05 02:28:24唐文東高陽艷孫予罕
物理化學學報 2010年5期
唐文東 高陽艷 魏 偉 孫予罕
(1中國科學院山西煤炭化學研究所,太原 030001; 2中國科學院研究生院,北京 100049; 3中國科學院上海高等研究院低碳能源研究中心,上海 201203)
唐文東1,2高陽艷1,2魏 偉1,*孫予罕1,3,*
(1中國科學院山西煤炭化學研究所,太原 030001;2中國科學院研究生院,北京 100049;3中國科學院上海高等研究院低碳能源研究中心,上海 201203)
運用VASP(Vienna ab-initio simulation package),采用基于密度泛函理論(DFT)的第一原理計算,研究了尿素在ZnO(101ˉ0)表面的吸附行為.計算結果表明:尿素分子在ZnO(101ˉ0)表面主要發生分子吸附,穩定的吸附產物通過尿素分子中的氮原子或氧原子與表面鋅原子之間的鍵合作用而形成,吸附能分別為-1.48和-1.41 eV;表面吸附的尿素分子也可以發生解離,生成表面吸附的異氰酸根、氨氣和一個表面羥基,吸附能為-1.66 eV.
吸附;尿素;ZnO;碳酸二甲酯;VASP
近年來,人們為了克服合成碳酸二甲酯傳統工藝的缺點,如有毒性、易爆炸性、反應工藝復雜和低轉化率等,發展了一種滿足“綠色化學”要求[1]的新型合成方法,即以尿素和甲醇為原料合成碳酸二甲酯[2-5](圖1).
為了提高反應的轉化率,人們研究了包括堿、有機錫化合物、金屬氧化物、含鋅化合物在內的一系列催化劑[2,6-15],結果表明,在這一系列催化劑中,ZnO由于其低毒性,高催化活性和易于分離的優點而被認為是最具潛力的催化劑[2,16].為了對該尿素甲醇化反應有一個清晰的認識,人們設計了許多實驗來研究反應的機理,并且提出了幾種機理模型[2,17-18].Wang等[2]認為,ZnO對第二步反應具有很高的活性,并且把這種高活性歸因于氧化鋅的酸堿性質.祁增忠等[17]則認為,在反應過程中,首先是氧化鋅吸附甲醇,吸附的甲醇再與尿素作用生成中間產物氨基甲酸甲酯,該中間產物再和另一分子的甲醇反應生成終產物碳酸二甲酯.Zhao等[18]則做了更進一步的研究:他們比較了分別從尿素和氨基甲酸甲酯為原料合成碳酸二甲酯的醇解反應,發現氧化鋅對第二條路線即氨基甲酸甲酯和甲醇合成碳酸二甲酯幾乎沒有催化活性,而它卻是第一條路線的決速步驟.因此,他們認為氧化鋅只是催化劑的前驅體,真正的催化劑是由氧化鋅轉變成的另外一種物質.通過對該物質的提取和表征,他們建議該物質的化學式為Zn(NH3)2(NCO)2,該物質通過三步過程生成:尿素首先分解成HNCO和NH3,接著HNCO和ZnO作用生成Zn(NCO)2,該物質再和NH3通過配合作用生成Zn(NH3)2(NCO)2.然而整個反應的機理仍然沒有搞清楚.
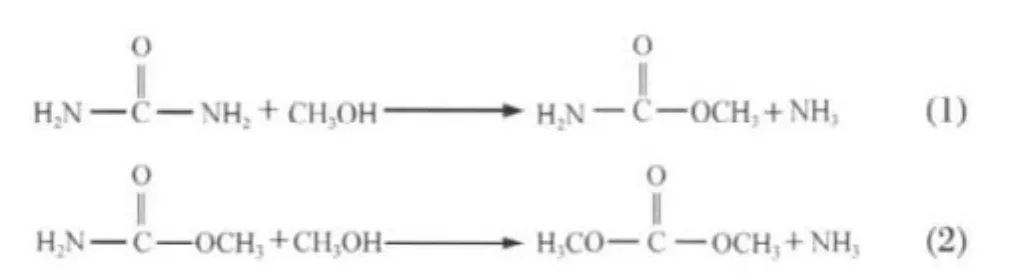
圖1 尿素和甲醇合成碳酸二甲酯的反應方程式Fig.1 Reactions of dimethyl carbonate(DMC) synthesis from urea and methanol
要認識非均相反應的機理,首先要認識非均相催化劑在反應過程中的催化行為,而考察反應分子在催化劑表面的吸附則是第一步.對于我們的反應體系,反應物為尿素和甲醇,催化劑為氧化鋅,由于Raj等[19]對甲醇在氧化鋅表面的吸附行為已做了相關研究,因此本文工作主要研究尿素在氧化鋅表面的吸附行為.這對認識整個反應過程的反應機理具有很重要的意義.
1 計算方法和模型
本文中的所有計算都是通過VASP(Vienna abinitio simulation package)[24]完成的.采用由Bl?chl[25], Kresse和Joubert[26]發展的綴加投影波函數(PAW)方法來描述電子-離子的相互作用(electron-ion interactions),交換相關能采用廣義梯度函數(GGA) PW91[27]進行計算,緊束縛芯電子采用PAW-GGA贗勢進行描述.所有計算中平面波截斷能(planewave basis cutoff energy)和布里淵區k點分別設置為350 eV和2×2×1,除了在模擬尿素時采用了5×5×5的布里淵區k點設置.經過結構優化的氧化鋅元胞的晶格參數為a=b=0.32493 nm,c=0.52054 nm,該結果和Sawada等[28]的實驗值,a=b=0.32490 nm,c= 0.52052 nm,非常吻合.表1中還列出了其他一些理論計算結果[29-32],均表明我們的結果與實驗值符合得比較好.
2 結果與討論
尿素可以在ZnO(101ˉ0)表面發生分子吸附和解離吸附.我們首先設計了尿素分子在氧化鋅表面發生分子吸附的可能構型,進行結構優化以確定最穩定的分子吸附模型,然后再以最穩定的分子吸附模型出發,進一步研究解離吸附.
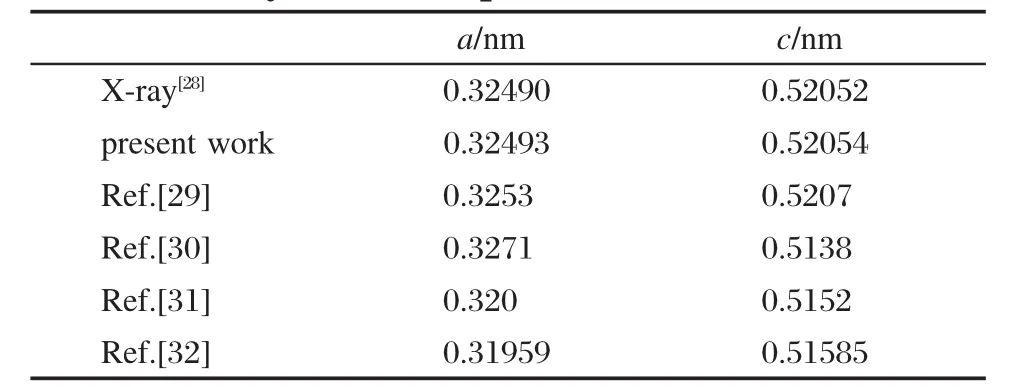
表1 ZnO元胞的晶格參數Table 1 Crystal lattice parameters of ZnO unit cell
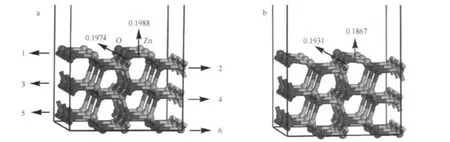
圖2 ZnO(1010)表面的超晶胞模型Fig.2 Supercell models of the ZnO(1010)surface(a)unrelaxed surface structure,(b)relaxed surface structure;The numbers 1 to 6 in Fig.1(a)represent the atomic layers 1 to 6,respectively. bond length in nm
本文中我們只計算了各種可能吸附構型的吸附能,以此來確定最穩定的吸附構型,并且期望能從中得到一些關于催化行為的信息.所有的吸附能由以下公式計算所得:
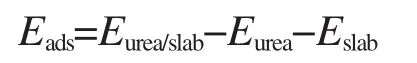
其中,Eurea/slab表示吸附復合物的總能,Eurea表示尿素分子的總能,Eslab則表示表面所在的超晶胞的總能.Eads為負值時就表明該吸附是一放熱過程.
2.1 分子吸附
所有可能的分子吸附構型的優化結構和其相應的吸附能列于圖3中.
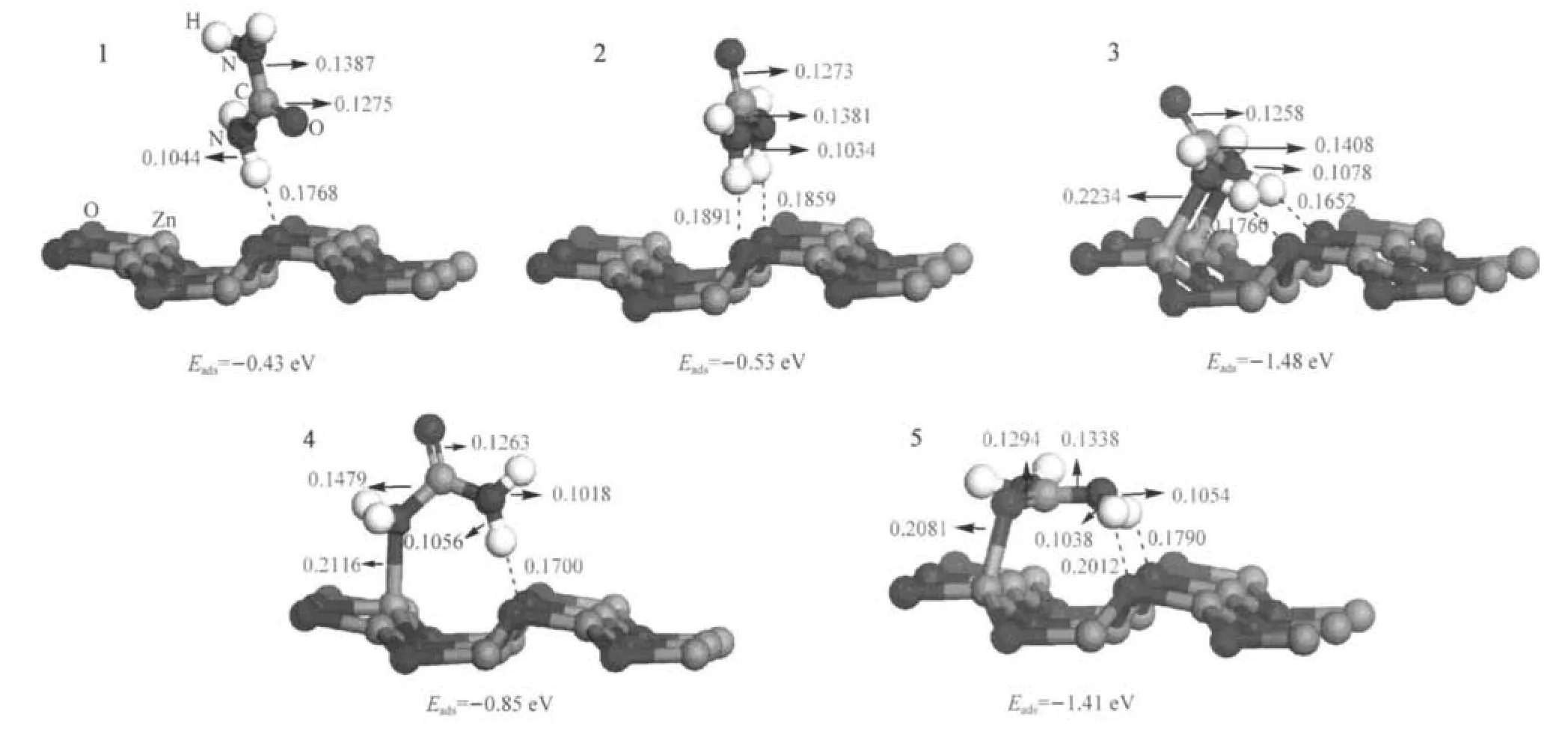
圖3 尿素在ZnO(1010)表面發生分子吸附的優化構型及其相應的吸附能Fig.3 Optimized structures and adsorption energies of the molecular adsorption modes of urea on the ZnO(1010)surfaceWe used the top two layers of the ZnO(1010)supercell for short.bond length in nm
計算結果指出,尿素分子傾向于通過其氮原子和表面的鋅原子發生鍵合作用而形成相對比較穩定的吸附復合物(圖3,構型3和4).在這兩種吸附構型中,和表面鋅原子發生作用的氮原子所在的C— N鍵變長,C=O鍵變短;構型3新形成兩個N—Zn鍵和兩個氫鍵;構型4中則只形成一個N—Zn和一個氫鍵.吸附的發生也使表面結構發生了變化,在這兩種構型中,表面的O1—Zn1和O2—Zn1鍵都有明顯的增長(此處的Zn1原子是表示和氮原子相連接的鋅原子),使得表面的鋅原子有從表面離開的趨勢.構型3中兩個N—Zn鍵和兩個氫鍵的形成使其比構型4更穩定,二者的吸附能分別為-1.48和-0.85 eV.
另一種比較穩定的吸附構型是尿素分子通過其羰基中的氧原子和表面鋅原子發生鍵合作用的情況.O—Zn鍵的生成減弱了C=O鍵的作用,C=O鍵鍵長變長;兩個氫鍵的形成也減弱相應的N—H鍵的作用,鍵長變長;表面結構也因吸附作用的發生而發生改變,表面的O1—Zn1和O2—Zn1都有所增長.在該構型中,由于分子和表面之間形成了一個O—Zn鍵和兩個氫鍵,使得該吸附構型也相對比較穩定,吸附能高達-1.41 eV.
從我們的計算結果來看,所有的分子吸附過程都是放熱反應.這表明,它們都是熱力學上有利的吸附構型,吸附能絕對值越大,其相應的吸附復合物就越穩定.因此,這些吸附復合物的穩定性和被吸附的機會具有相同的順序:即3>5>4>2>1.如果把吸附能作為產率的粗略近似,根據吸附能Eads之間較大的差異,通過假設平衡的粗略估算可以得知,在ZnO(101ˉ0)表面上,吸附復合物1、2和4幾乎不會存在,而吸附復合物3和5則是最多的.這就意味著在整個分子吸附過程中,吸附復合物3和5是主要的分子吸附產物.
2.2 解離吸附
在表面原子的作用下,表面吸附的分子可以發生解離作用.我們以2.1節中得到的兩種主要分子吸附產物為起始點,進一步研究尿素在表面的解離吸附情況.解離吸附的優化構型及相應的吸附能列于圖4中.
在我們的結果中只得到了一種解離吸附復合物,來源于分子吸附復合物3.我們用同樣的符號標記解離吸附復合物以表明它們的來源(即分子吸附復合物).該解離吸附復合物來自于尿素分子中的N—H鍵的斷裂,形成了一個表面羥基和一個H2NCONH自由基,該自由基的兩個氮原子和表面的鋅原子鍵合在一起.通過對吸附能的比較,我們發現,解離吸附復合物沒有其相應的分子吸附復合物穩定.解離吸附復合物的吸附能為-1.37 eV,而其相應的分子吸附復合物的吸附能為-1.48 eV,解離需吸熱0.11 eV.這表明,尿素分子在ZnO(101ˉ0)表面傾向于發生分子吸附.
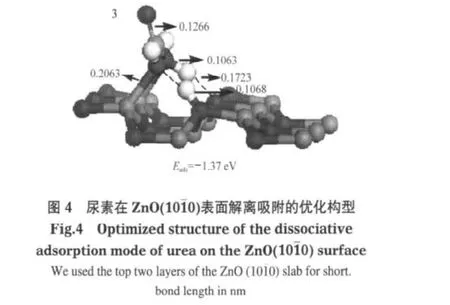

研究發現,初次解離吸附產物可以發生進一步的解離.計算結果表明,解離吸附復合物3可以通過C—N鍵的斷裂,生成表面吸附的氨氣、異氰酸根和一個表面羥基,形成更穩定的二次解離吸附復合物3(Eads=-1.66 eV),放出0.29 eV的能量(圖5).
3 結論
致謝: 感謝中國科學院山西煤炭化學研究所霍春芳副研究員的有益討論和幫助.
1 Tundo,P.;Selva,M.Accounts Chem.Res.,2002,35(9):706
2 Wang,M.H.;Zhao,N.;Wei,W.;Sun,Y.H.Ind.Eng.Chem.Res., 2005,44(19):7596
3 Yang,B.L.;Wang,D.P.;Lin,H.Y.;Sun,J.J.;Wang,X.P.Catal. Commun.,2006,7(7):472
4 Wang,M.H.;Wang,H.;Zhao,N.;Wei,W.;Sun,Y.H.Catal. Commun.,2006,7(1):6
5 Sun,J.J.;Yang,B.L.;Wang,X.P.;Wang,D.P.;Lin,H.Y. J.Mol.Catal.A-Chem.,2005,239(1-2):82
6 Cho,T.;Tamura,T.;Cho,T.;Suzuki,K.Process for preparing dialkyl carbonates:US,5534649[P].1996-10-15
7 Lin,H.Y.;Yang,B.L.;Sun,J.J.;Wang,X.P.;Wang,D.P. Chem.Eng.J.,2004,103(1-3):21
8 Saleh,R.Y.;Michaelson,R.C.;Suciu,E.N.;Kuhlmann,B. Process for manufacturing dialkyl carbonate from urea and alcohol: US,5565603[P].1996-10-15
9 Ryu,J.Y.;Gelbein,A.P.Process and catalyst for making dialkyl carbonates:US,6392078[P].2002-05-21
10 Wu,C.C.;Zhao,X.Q.;Wang,Y.J.Catal.Commun.,2005,6(10): 694
11 Bhanage,B.M.;Fujita,S.;Ikushima,Y.;Arai,M.Green Chem., 2003,5(4):429
12 Li,Q.B.;Zhang,W.Y.;Zhao,N.;Wei,W.;Sun,Y.H.Catal. Today,2006,115(1-4):111
13 Zhao,X.Q.;Zhang,Y.;Wang,Y.J.Ind.Eng.Chem.Res.,2004, 43(15):4038
14 Suciu,E.N.;Kuhlmann,B.;Knudsen,G.A.;Michaelson,R.C. J.Organomet.Chem.,1998,556(1-2):41
15 Wang,D.P.;Yang,B.L.;Zhai,X.W.;Zhou,L.G.Fuel Process. Technol.,2007,88(8):807
16 Zhao,W.B.;Wang,F.;Peng,W.C.;Zhao,N.;Li,J.P.;Xiao,F. K.;Wei,W.;Sun,Y.H.Ind.Eng.Chem.Res.,2008,47(16):5913
17 Qi,Z.Z.;Wang,H.B.;Xia,D.K.Industrial Catalysis,2006,14 (1):26 [祁增忠,王洪波,夏代寬.工業催化,2006,14(1):26]
18 Zhao,W.B.;Peng,W.C.;Wang,D.F.;Zhao,N.;Li,J.P.;Xiao,F. K.;Wei,W.;Sun,Y.H.Catal.Commun.,2009,10(5):655
19 Raj,G.S.P.;Horia,M.J.Catal.,2008,254(2):325
20 Jan,W.;Jacek,P.Solid State Commun.,2008,146(7-8):324
21 Gay,R.R.;Nodine,M.H.;Henrich,V.E.;Zeiger,H.J.;Solomon, E.I.J.Am.Chem.Soc.,1980,102(22):6752
22 Marana,N.L.;Longo,V.M.;Longo,E.;Martins,J.B.L.; Sambrano,J.R.J.Phys.Chem.A,2008,112(38):8958
23 Sarano,D.;Spoto,G.;Bordiga,S.;Zecchina,A.;Lamberti,C.Surf. Sci.,1992,276(1-3):281
24 Kresse,G.;Furthmuller,J.Comput.Mater.Sci.,1996,6(1):15
25 Bl?chl,P.E.Phys.Rev.B,1994,50(24):17953
26 Kresse,G.;Joubert,D.Phys.Rev.B,1999,59(3):1758
27 Perdew,J.P.;Chevary,J.A.;Vosko,S.H.;Jackson,K.A.; Pederson,M.R.;Singh,D.J.;Fiolhais,C.Phys.Rev.B,1992,46 (11):6671
28 Sawada,H.;Wang,R.P.;Sleight,A.W.J.Solid State Chem., 1996,122(1):148
29 Beltran,A.;Andres,J.;Calatayud,M.;Martins,J.B.L.Chem. Phys.Lett.,2001,338(4-6):224
30 Binks,D.J.;Grimes,R.W.J.Am.Ceram.Soc.,1993,76(9):2370
31 Gopal,P.;Spaldin,N.A.J.Electron.Mater.,2006,35(4):538
32 Cooke,D.J.;Arnaud,M.;Stephen,C.P.J.Phys.Chem.B,2006, 110(15):7985
33 Nyberg,M.;Nygren,M.A.;Pettersson,L.G.M.;David,H.G.; Rohl,A.L.J.Phys.Chem.,1996,100(21):9054
34 Wander,A.;Harrison,N.M.Surf.Sci.,2000,457(1-2):342
October 19,2009;Revised:February 5,2010;Published on Web:March 19,2010.
Adsorption of Urea onto a ZnO()Surface
TANG Wen-Dong1,2GAO Yang-Yan1,2WEI Wei1,*SUN Yu-Han1,3,*
(1Institute of Coal Chemistry,Chinese Academy of Sciences,Taiyuan 030001,P.R.China;2Graduate University of Chinese Academy of Sciences,Beijing 100049,P.R.China;3Low Carbon Energy Center,Shanghai Advanced Research Institute,Chinese Academy of Sciences,Shanghai 201203,P.R.China)
First-principles calculations based on density functional theory(DFT)were used to investigate the adsorption of urea onto a nonpolar ZnO(101ˉ0)surface with the VASP(Vienna ab-initio simulation package)code.The calculation results indicated that urea was favorably adsorbed onto the ZnO(101ˉ0)surface molecularly,and that stable adsorption products were formed through the reaction between nitrogen atom or oxygen atom from urea and zinc atom on the surface.The adsorption energy was-1.48 and-1.41 eV,respectively.The adsorbed urea can dissociate to form an isocyanic radical,an ammonia molecule,and a surface hydroxyl,all of which adsorb onto the surface.The adsorption energy was-1.66 eV.
Adsorption;Urea;ZnO;Dimethyl carbonate;VASP
[Article] www.whxb.pku.edu.cn
*Corresponding authors.Email:weiwei@sxicc.ac.cn,yhsun@sxicc.ac.cn;Tel:+86-351-4053801.
The project was supported by the Key Technology R&D Program for 11th Five-Year Plan,China(07ZCU11691)and the Key Project of Knowledge Innovation Program of Chinese Academy of Sciences(08YCA21691).
“十一五”國家科技支撐計劃重大項目(07ZCU11691)和中國科學院知識創新重要方向性項目(08YCA21691)資助
O641
