基于粗粒化模型對有機溶劑的分子動力學模擬
2011-11-30 10:50:42許佩軍唐媛媛張知博
物理化學學報 2011年8期
關鍵詞:模型
許佩軍 唐媛媛,2 張 靜 張知博 王 昆
邵 穎3 沈虎峻2,* 毛英臣1,*
(1遼寧師范大學物理與電子技術學院,遼寧大連116029;2中國科學院大連化學物理研究所,遼寧大連116023; 3大連海事大學物理系,遼寧大連116026)
基于粗粒化模型對有機溶劑的分子動力學模擬
許佩軍1唐媛媛1,2張 靜1張知博1王 昆1
邵 穎3沈虎峻2,*毛英臣1,*
(1遼寧師范大學物理與電子技術學院,遼寧大連116029;2中國科學院大連化學物理研究所,遼寧大連116023;3大連海事大學物理系,遼寧大連116026)
在基于Boltzmann分布對四種基本構象進行Monte Carlo取樣后,通過與全原子模型的范德華勢比較得到了Gay-Berne(GB)參數.又在對用量化計算得到的分子體系的電勢進行電荷、偶極矩和四極矩的擬合后,得到了電多極展開勢(EMP)參數.利用得到的粗粒化參數,基于粗粒化模型,對CHCl3及四氫呋喃(THF)兩種有機溶劑進行了分子動力學模擬(MDS),并將結果同全原子模擬進行了比較.計算結果表明用粗粒化模型從整體上能重復全原子模型的模擬結果,但在某些細節的計算與全原子模型有偏差,其原因可能是目前工作僅考慮了單位點情況,為此今后在對具有復雜結構的分子進行粗粒化模擬時還應考慮合理放置及增加相互作用位點.
粗粒化模型;Gay-Berne勢;電多極展開勢;徑向分布函數;分子動力學模擬
1 引言
分子動力學模擬(MDS)已經成為了研究復雜分子體系的重要工具,盡管一些計算方法和技術手段已經取得了突破,1-3但當前基于全原子模型對某些較大體系的動力學模擬時間尺度仍然非常短,并不能滿足與實驗比較的迫切需要.4-6已知在利用經驗力場進行的全原子分子動力學模擬主要包括兩部分:一是對分子的構象進行統計分析;二是結合相應的構象分析體系的能量及受力情況.7-9而對某一分子體系,由于非鍵結的范德華相互作用項和靜電相互作用項的數目遠遠大于鍵長、鍵角和二面角相互作用項的數目,因此對范德華相互作用和靜電相互作用的計算將要耗費大量的計算時間.為了簡化對這兩種相互作用的計算,研究人員已提出了多種粗粒化模型(CG模型).10-14CG模型的主要特點在于它將研究體系中的分子或分子的某一部分近似看成一個原子團簇,該原子團簇內部信息(鍵長、鍵角、二面角等)被屏蔽,即該團簇被處理為沒有內部結構的“粒子”,該粒子對應于全原子模型中的單個原子.而在利用CG模型進行分子動力學模擬之前還必須獲得描述這些粒子間范德華相互作用和靜電相互作用的力場參數,應當指出這一部分工作也是構建CG模型的關鍵部分.CG模型結合了日益發展的計算機水平,使得對更大更復雜體系的長時模擬成為可能.
該方法的主要缺陷就在于它的計算精度要低于基于全原子模型的分子動力學模擬.10此外還需指出的是大多數的CG模型僅對某些特殊體系適用,而不能應用于其他體系,即模型的可移植性有待提高.產生這一問題的主要原因在于多數CG模型將粗粒化粒子看成是球形,這樣導致了這些模型沒能更好地描述分子間的范德華相互作用,以及未能對由此而導致的電勢空間分布改變的靜電相互作用作出更好的描述.為了克服這些缺陷,改進CG模型,Golubkov和Ren14提出了在計算粗粒化粒子間相互作用時,必須考慮由粗粒化粒子的形狀而引起的Gay-Berne(GB)效應及電多極矩效應,這些改進在他們對水、苯及甲醇三種構象相對簡單的分子體系的動力學模擬中得到了較理想體現.此外也應認識到的是他們工作的基石就在于較好地擬合得到了描述上述三種分子的粗粒化力場參數.為了檢驗如何利用CG模型對復雜分子體系進行高效計算,本文采用類似方法對CHCl3及四氫呋喃(THF)分子的兩種構象在相對復雜的有機溶劑中進行了分子動力學模擬.
2 計算與模擬方法
2.1 描述非球形分子的Gay-Berne勢和電多極展開勢(EMP)
任何一個好的模型必須具備兩個主要特征,一方面數學上要簡單并易于處理,另一方面能夠對模擬體系做較好的物理描述.基于這兩方面的考慮, Berne和Pechukas15近似地將分子視為繞其主軸旋轉的橢球體,得到了由高斯函數描述的經驗勢.在對非球形分子的分子模擬中,短距離吸引和排斥相互作用可通過如下方法來完成,即在分子內定義多個位點,每兩個位點間的范德華相互作用勢由Lennard-Jones勢描述.需指出的是對于較大分子,計算勢的時間會隨位點數目的平方而遞增,從而使得計算效率大為降低.為了解決這一問題,Gay和Berne16對描述非球形分子間的短距離排斥和相互吸引作用的高斯重疊勢進行了修正,把分子處理成軟的、單軸的橢球體,把GB位點放置在橢球體的質心上,用單位點勢替代多位點勢,這樣就得到了GB勢.在慣性系中,橢球體在位形空間中由其質心坐標和三個歐拉角描述,對應于由一套GB參數來描述.目前,GB勢在描述液晶體系中得到了廣泛應用.17-20為了更好地控制GB勢的軟硬度,Brene和Pechukas建議增加參數來控制經驗勢的柔性,由此Kabadi等21,22引入了dw參數.
兩個粗粒化粒子間的GB勢可表示為


其中

其中l和d分別描述分子的長和寬,這樣分子的形狀就可以用任意的棒狀、盤狀或者球狀來表示.
(1)式中的阱深參數取如下形式

其中μ和ν均為可調指數,借鑒Golubkov和Ren14的工作,本文分別將其取值為2.0和1.0.而ε1和ε2可分別表示為

(9)式中的χ?和α?分別取如下形式

其中,ε0描述了交叉結構的阱深,εE和εS分別表示尾對尾和邊對邊結構的阱深.當兩個分子相同或者其中一個分子是球形分子時,勢函數用Berne和Pechukas的原始形式表示.如果兩個分子都是球形的,勢函數將進一步簡化為Lennard-Jones勢.
長距離靜電相互作用可以由電多極展開勢來表示.在大于分子尺寸的距離處,分子的電勢可準確地通過對其質心的電多極展開勢來表示23
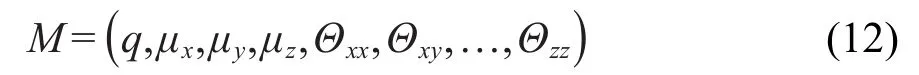
其中q、μ、Θ分別表示電荷、偶極矩和四極矩.電多極勢可表示為電荷-電荷相互作用勢、電荷-偶極相互作用勢、電荷-四極相互作用勢等項之和.本文在分子的質心上放置了一個電多極展開位點,同時選取了和GB位點相同的慣性系.
兩個多極位點間的相互作用可通過電多極展開勢表示為

其矩陣形式可參見文獻.14,24本文對靜電相互作用截斷值的處理方法,采用了TINKER軟件的標準方法.25-27
此外還需指出的是,考慮到由于粗粒化粒子具有各向異性的形狀而使得在進入排斥的范德華相互作用區域前電多極展開方法將給出不正確的相互作用勢,類似于文獻,14,28本文同樣加入了一個阻尼函數以確保對短距離多極相互作用勢的精確描述.
2.2 粗粒化模型力場參數的確定
2.2.1 Gay-Berne(GB)參數的擬合
本文利用粗粒化模型進行分子模擬的前提是要獲得GB-EMP模型的參數,為此我們首先對研究體系進行了合理取樣選取,然后利用描述有機分子的AMBER力場(GAFF),29,30通過與全原子的范德華勢進行比較后擬合得到了GB參數.選取GAFF力場是由于它的函數形式簡單,原子類型有限,還可利用實驗和理論模型計算獲得力常數和部分原子電荷.在研究中,我們發現構象的選取會直接影響GB參數的擬合,進而決定GB勢是否能準確地模擬全原子的范德華勢,因此選擇可以反映真實分子液體環境的構象是十分重要的.由于在實際的溶液環境中,面對面(或者尾對尾)、邊對邊、T形和交叉結構這四種構象出現的幾率相對較大,17,19另外考慮到在真實的液體環境中分子的構象趨于取能量最低的構象,所以本文在篩選用于擬合GB參數的構象時,對上述四種構象分別基于Boltzmann分布采用Monte Carlo方法獲得到分子的構象集.這樣才可確保在一定的能量范圍內,構象數目隨能量的升高而減少.表1給出了CHCl3和THF這兩種分子的四種基本構象.

表1 氯仿和THF分子的四種基本構象Table 1 Four reference configurations of CHCl3and THF molecules
圖1及圖2給出了氯仿和THF分子的四種基本構象在各能量區間內的構象數目.從圖中我們可以清楚地看到所選的分子構象在較低的能量范圍內較多,且構象數隨著能量的升高而逐漸減小.

表2 氯仿和THF分子的GB參數Table 2 GB parameters of CHCl3and THF molecules
在參數擬合的具體過程中,5個自由參數(l,d, ε0,εE/εS,dw)由遺傳算法擬合得到.表2給出用上述方案擬合得到的氯仿和THF分子的GB參數.
2.2.2 EMP的參數擬合
在擬合粗粒化模型的EMP參數時,本文首先應用Gaussian 03軟件包31的MP2方法采用6-311G** (2d,2p)機組計算得到了兩種分子的電勢,然后利用GDMA程序32對電勢進行電荷、偶極和四極的擬合,最后得到了兩種分子粗粒化模型的EMP參數.表3中給出了氯仿和THF的EMP參數.


2.3 模擬體系與動力學模擬方法
為了檢驗本文對氯仿和THF分子粗粒化模型GB-EMP參數的擬合,我們利用粗粒化模型對兩種有機分子溶劑進行了粗粒化的分子動力學模擬.為了在計算的過程中可任意地在粗粒化模型和全原子模型間進行轉換,方便容易地比較CG模型和全原子模型的結果,故本文在計算中采用了TINKER軟件包.此外還基于如下考慮,即TINKER也包含了對粗粒化粒子GB-EMP能量、力和力矩部分的計算,并可在慣性系中非常容易地整合GB和EMP兩部分的計算結果.
氯仿和THF有機溶劑的立方體盒子由Materials Studio(MS)5.0軟件生成,其邊長均為3 nm.對GB和EMP兩部分的截斷半徑都取為1.2 nm(這考慮到在通常情況下,要求截斷半徑值不大于盒子邊長的一半).CG模型和全原子模型的分子動力學模擬都是采用NVT系綜在室溫(298 K)下進行的.基于全原子模型的分子動力學模擬的計算步長取為1 fs.文獻14對考慮了GB-EMP效應的CG模型的時間步長作了實驗,推薦CG模型的計算步長可取5-20 fs.考慮到由于自由度的減小,以及分子內高頻運動的“屏蔽”,在本文我們取時間步長為5 fs.在對運動方程進行數值化處理時,兩種模型的計算都采用了TINKER推薦的BERMAN積分算法.25
3 結果與討論
3.1 對范德華相互作用的分析
在圖3中,我們比較了分別利用CG模型和全原子模型計算得到的氯仿分子的范德華相互作用勢,可以看到CG模型結果與全原子模型結果總體符合得較好.對面對面及交叉結構,兩種方法得到的作用曲線幾乎完全相同;而對于邊對邊及T型構象,可以看出相比于全原子模型,粗粒化模型得到了較小的阱深和距離參數,這一現象對T型構象尤為明顯.此外,基于粗粒化模型對四種構象的計算結果整體趨勢較好地符合了Gay及Berne16的結論.
通過將分子構象的取樣圖(圖1)與范德華相互作用勢能曲線圖(圖3)進行比較,可以發現分子構象數最多的能量區域正好對應于范德華相互作用勢能曲線圖的最低點區域,這也印證了分子構象都趨于位于能量最低的構象狀態的結論.19
圖4給出了基于兩種模型對THF分子范德華相互作用的比較.從圖中可以看出,相比于結構簡單的氯仿分子,對THF分子進行粗粒化模型的計算結果與全原子結果有較大的偏差.這表現在對應于四種基本構象的阱深參數和距離參數都一致性地小于全原子模型的計算結果.從圖3和圖4中,我們也觀察到兩種分子的交叉結構和邊對邊結構的范德華作用曲線非常接近,幾乎重合,這一現象很好地支持了文獻16的結論.在對粗粒化參數做多次擬合后,計算結果仍然如此,由此我們認為出現這一現象的主要原因是我們僅考慮了單位點情況.這也建議在用粗粒化模型對復雜結構的分子來計算分子間范德華相互作用時應考慮增加相互作用位點.

圖3 基于粗粒化模型和全原子模型對氯仿分子的范德華相互作用曲線的比較Fig.3 Comparison of van der Waals potential of CHCl3 based on the CG model and the all-atom model The curves of four configurations,namely face to face(lines 1 and2),side by side(lines 5 and 6),T-shape(lines 3 and 4),and cross shape(lines 7 and 8),are also displayed,respectively.

圖4 基于粗粒化模型和全原子模型對THF分子的范德華相互作用曲線的比較Fig.4 Comparison of van der Waals potential of THF based on the CG model and the all-atom modelThe curves of four configurations,namely face to face(lines 1 and 2),side by side(lines 5 and 6),T-shape(lines 3 and 4)and cross shape(lines 7 and 8),are also displayed,respectively.The results of the CG model are represented with solid lines,and the dash lines are the results of the all-atom model.
考慮到粗粒化模型的普遍特征,10在目前階段的工作中,要進一步檢驗粗粒化模型的計算效率,還需結合其他物理量,如溶劑的徑向分布曲線等來做進一步研究.
3.2 徑向分布函數的比較分析
需要指出,為了比較兩種模型得到的徑向分布函數曲線,首先需要把用粗粒化模型得到的CG軌跡轉換為全原子軌跡.圖5給出了氯仿分子的徑向分布函數曲線.從圖中可以看出,盡管時間步長增大為5 fs,粗粒化的分子動力學模擬仍能給出一條穩定的軌跡,這說明CG模型能夠較好地重復全原子的模擬結果.與全原子模型相比,CG模型的徑向分布函數曲線起伏較少,這符合粗粒化模型的基本特征.從C-H徑向分布曲線可看出,在0.65 nm處CG模型得到一波峰,此外從Cl-Cl及H-Cl徑向分布曲線還可看出CG模型不能重現0.30 nm處的波峰,但當距離較大時,CG模型和全原子模型的結果符合得較好.在距離較小處,兩者的結果符合得較差,其原因可能與EMP相互作用位點的放置有關.在本文中,與GB相互作用位點的放置相同, EMP作用位點放置在粗粒化粒子的質心上,這樣就造成了對電多極展開勢空間分布描述的偏差,從而影響了對電負性較強元素(Cl元素)的相關徑向分布函數曲線的模擬.而我們注意到Golubkov和Ren等33在最近實驗工作中把EMP作用位點放到了其他位置上,相比于他們之前工作,14這樣更好地模擬了所研究體系的相關性質.這也提示在以后的工作中,應針對分子的具體結構,考慮把EMP位點放在其他位置(如C原子上)而不是分子的質心,或者考慮增加額外的相互作用位點.
圖6給出了THF溶劑的徑向分布函數曲線.可以看出CG模型能較好地重復全原子的模擬結果,然而必須指出的是,與兩種方法得到的范德華相互作用曲線的較大差異不同,兩種模型的模擬結果符合得較好,沒有出現類似于氯仿的徑向分布曲線中的較大差異.鑒于分子結構的差異,相比于將EMP相互位點放置在氯仿粗粒化粒子的質心上,將該位點放置在THF粗粒化粒子的質心上,目前我們認為是一種可以接受的處理方式.對于CG模型仍然不能較好地重復全原子模型在0.30 nm處的較小起伏,我們認為考慮增加相互作用位點或者適當地調整EMP作用位點的位置會提高粗粒化模型的計算精度.

圖5 對基于兩種模型得到的氯仿溶劑的徑向分布函數(RDF)曲線的比較Fig.5 Comparison of radial distribution functions(RDF)of CHCl3solvent based on the two models(a)C-H,(b)Cl-Cl,(c)H-Cl;Theresultsof theCG modelarerepresentedwithsolidlines,andthedash linesaretheresultsof theall-atommodel.

圖6 對基于兩種模型得到的THF溶劑的徑向分布函數(RDF)曲線的比較Fig.6 Comparison of radial distribution functions(RDF)of THF solvent based on the two models(a)C-H,(b)H-H,(c)O-H;The results of the CG model are represented with solid lines,and the dash lines are the results of the all-atom model.
通過CG模型進行分子動力學模擬的主要優點是可以在保證一定精度的條件下較大幅度地提高計算的效率,這主要是由于用粗粒化的GB-EMP相互作用代替了多對原子間的相互作用,使得自由度數得到了較大地減少.此外一重要因素便是由于忽略了鍵長、鍵角和二面角的高頻運動,而使得模擬的時間步長增大了.
還需指出的是,由于在處理Gay-Berne效應中引入的dw參數較好地控制了GB勢的柔性,所以用本文中的CG模型還可用來模擬更大的復雜分子體系.此外在考慮了能更精確地描述靜電相互作用的電多極勢效應后,使得基于CG模型模擬那些帶高電量的生物大分子體系成為可能.
4 結論
為了精確描述非球形分子間的相互作用,考慮了Gay-Berne效應和電多極勢效應,基于粗粒化模型對氯仿和THF兩種有機溶劑進行了分子動力學模擬,并同全原子模擬結果進行了比較.為此本文首先對研究體系的分子構象進行了合理取樣,然后利用描述有機分子的GAFF力場,通過與全原子的范德華勢進行比較后,用遺傳算法擬合得到了GB參數.在擬合GB參數的過程中,我們也注意到構象選取對計算結果的影響,針對CHCl3和THF兩種有機溶劑中面對面、邊對邊、T型及交叉結構這四種構象出現幾率相對較大,及分子構象都趨于位于能量最低的構象狀態的特征,利用Monte Carlo方法基于Boltzmann分布得到了擬合參數的構象集,最后利用遺傳算法得到了描述粗粒化粒子間相互作用的GB參數.結合利用量化計算得到的EMP參數,我們就獲得到了精確描述兩種有機分子的GB-EMP參數.利用得到的粗粒化參數,基于粗粒化模型對范德華相互作用曲線和徑向分布函數曲線進行的計算表明粗粒化模型能以較高的效率從整體上重復全原子的計算結果,但是在某些細節方面,粗粒化模型不能很好地符合全原子的分子動力學模擬結果,這主要與目前工作描述相互作用的位點較少及相互作用位點放置簡單有關.為此我們應在下一步工作中增加相互作用位點,并應適當地調整GB位點和EMP位點的具體位置.
(1) Dror,R.O.;Jensen,M.?.;Borhani,D.W.;Shaw,D.E.J.Gen. Physiol.2010,135,555.
(2) Karplus,M.;McCammon,J.A.Nature Struct.Biol.2002,9, 646.
(3) Shaw,D.E.;Maragakis,P.;Lindorff-Larsen,K.;Piana,S.;Dror, R.O.;Eastwood,M.P.;Bank,J.A.;Jumper,J.M.;Salmon,J. K.;Shan,Y.B.;Wriggers,W.Science 2010,330,341.
(4) van Gunsteren,W.F.;Bakowies,D.;Baron,R.;Chandrasekhar, I.;Christen,M.;Daura,X.;Gee,P.;Geerke,D.P.;Gl?ttli,A.; Hünenberger,P.H.;Kastenholz,M.A.;Oostenbrink,C.; Schenk,M.;Trzesniak,D.;van der Vegt,N.F.A.;Yu,H.B.B. Angew.Chem.Int.Edit.2006,45,4064.
(5) Klepeis,J.L.;Lindorff-Larsen,K.;Dror,R.O.;Shaw,D.E. Curr.Opin.Struct.Biol.2009,19,120.
(6) Freddolino,P.L.;Harrison,C.B.;Liu,Y.X.;Schulten,K. Nature Phys.2010,6,751.
(7)Xu,X.J.;Hou,T.J.;Qiao,X.B.;Zhang,W.Computer Aided Drug Design;Chemical Industry Press:Beijing,2004;pp 169-172.[徐筱杰,侯廷軍,喬學斌,章 威.計算機輔助藥物分子設計.北京:化學工業出版社,2004:169-172.]
(8) Leach,A.P.Molecular Modeling,Principles and Application; Person Education Limited:England,2001;pp 165-245.
(9) Schlick,T.Molecular Modeling and Simulation:An Interdisciplinary Guide,2nd ed.;Springer:New York,2010; pp 265-343.
(10) Voth,G.A.Coarse-Graining of Condensed Phase and Biomolecular Systems;CRC Press:England,2009.
(11) Chu,J.W.;Izvekov,S.;Voth,G.A.Mol.Sim.2006,32,211.
(12) Marrink,S.J.;de Vries,A.H.;Mark,A.E.J.Phys.Chem.B 2004,108,750.
(13) Tozzini,V.Curr.Opin.Struc.Biol.2005,15,114.
(14) Golubkov,P.A.;Ren,P.Y.J.Chem.Phys.2006,125,064103.
(15) Berne,B.J.;Pechukas,P.J.Chem.Phys.1972,56,4213.
(16) Gay,J.G.;Berne,B.J.J.Chem.Phys.1981,74,3316.
(17) Cleaver,D.J.;Care,C.M.;Allen,M.P.;Neal,M.P.Phys.Rev. E 1996,54,559.
(18) Wilson,M.R.J.Chem.Phys.1997,107,8654.
(19) Care,C.M.;Cleaver,D.J.Rep.Prog.Phys.2005,68,2665.
(20) Paramonov,L.;Yaliraki,S.N.J.Chem.Phys.2005,123, 194111.
(21) Kabadi,V.N.;Steele,W.A.Ber.Bunsenges.Phys.Chem.1985, 89,2.
(22) Kabadi,V.N.Ber.Bunsenges.Phys.Chem.1986,90,327.
(23) Jackson,J.D.Classical Electrodynamics,3rd ed;John Wiley& Sons Inc.:New York,1999;pp 145-150.
(24) Applequist,J.J.Phys.A 1989,22,4303.
(25) Ponder,J.W.TINKER Molecular Modeling,Package 5.1; Washington University Medical School.
(26) Darden,T.;York,D.;Pedersen,L.G.J.Chem.Phys.1993,98, 10089.
(27) Sagui,C.;Pedersen,L.G.;Darden,T.A.J.Chem.Phys.2004, 120,73.
(28) Ren,P.Y.;Ponder,J.W.J.Phys.Chem.B 2003,107,5933.
(29)Wang,J.M.;Wolf,R.M.;Caldwell,W.J.;Kollman,P.A.;Case, D.A.J.Comput.Chem.2004,25,1157.
(30)Yang,L.J.;Tan,C.H.;Hsieh,M.J.;Wang,J.M.;Duan,Y.; Cieplak,P.;Caldwell,W.J.;Kollman,P.A.;Luo,R.J.Phys. Chem.B 2006,110,13166.
(31) Frisch,M.J.;Trucks,G.W.;Schlegel,H.B.;et al.Gaussian 03, RevisionA.01;Gaussian Inc.:Pittsburgh,PA,2003.
(32) Stone,A.J.J.Chem.Theory Comput.2005,1,1128.
(33)Golubkov,P.A.;Wu,J.C.;Ren,P.Y.Phys.Chem.Chem.Phys. 2008,10,2050.
Febraury 28,2011;Revised:May 9,2011;Published on Web:June 14,2011.
Molecular Dynamics Simulation of Organic Solvents Based on the Coarse-Grained Model
XU Pei-Jun1TANG Yuan-Yuan1,2ZHANG Jing1ZHANG Zhi-Bo1WANG Kun1SHAO Ying3SHEN Hu-Jun2,*MAO Ying-Chen1,*
(1School of Physics and Electronic Technology,Liaoning Normal University,Dalian 116029,Liaoning Province,P.R.China;
2Dalian Institute of Chemical Physics,Chinese Academy of Sciences,Dalian 116023,Liaoning Province,P.R.China;
3Department of Physics,Dalian Maritime University,Dalian 116026,Liaoning Province,P.R.China)
To obtain Gay-Berne(GB)parameters,we carried out Monte Carlo sampling of four reference configurations based on the Boltzmann distribution.After comparing with the van der Waals potential within the all-atom model we obtained the GB parameters.Also by fitting the charge,dipole,and quadrupole with the electric potential obtained from quantum chemical computations with Gaussian 03 we obtained the electric multipole potential(EMP)parameters.With the GB-EMP parameters we then carried out molecular dynamics simulations(MDS)for CHCl3and tetrahydrofuran(THF)based on the coarsegrained(CG)model.Compared with the all-atom model,the CG model can reproduce the simulation results on the whole,but there are some deviations in the simulations in some details.The reason is that we only take one interaction site into account in this work.Therefore,for more complicated molecules it is necessary to take the placement of the interaction sites into account.Additionally,the multi-sites situation is also considered in the MDS within the frame of the coarse-grained model.
Coarse-grained model;Gay-Berne potential;Electric multipole potential;Radial distribution function;Molecular dynamics simulation
O645;O641
*Corresponding authors.MAO Ying-Chen,Email:myc@lnnu.edu.cn;Tel:+86-411-82158367.SHEN Hu-Jun,Email:hshen@dicp.ac.cn; Tel:+86-411-84379875.
The project was supported by the Fundamental Research Funds for the Central Universities,China(2009QN069).
中央高校專項資金優秀青年教師基金(2009QN069)資助項目
猜你喜歡
童話王國·奇妙邏輯推理(2024年5期)2024-06-19 16:03:38
網絡安全與數據管理(2022年1期)2022-08-29 03:15:20
導航定位學報(2022年4期)2022-08-15 08:27:00
中學生數理化·中考版(2022年8期)2022-06-14 06:55:24
新世紀智能(數學備考)(2021年9期)2021-11-24 01:14:36
成都醫學院學報(2021年2期)2021-07-19 08:35:14
新世紀智能(數學備考)(2020年9期)2021-01-04 00:25:14
中學生數理化·七年級數學人教版(2020年10期)2020-11-26 08:24:50
數學物理學報(2020年2期)2020-06-02 11:29:24
光學精密工程(2016年6期)2016-11-07 09:07:19

- 物理化學學報的其它文章
- Micellization Behavior of an Amphiphilic Drug Promethazine Hydrochloride-Surfactant System in an Aqueous Medium
- Synthesis of a Novel Thiadiazine Derivative and Electrochemical Properties for Pb2+Transfer across Water/1,2-Dichloroethane Interface
- YLuAG:Ce粉體的發光及閃爍特征:制備方法及缺陷效應
- 一種可作為FCC基質的新型改性鎂鋁尖晶石材料
- 納米碳纖維載鉑作為質子交換膜燃料電池陽極催化劑
- 乙烯基噻吩共軛螺噁嗪化合物的密度泛函理論研究