6-甲基-1-茚酮的光敏化合成與純化
2012-06-15 09:10:32王帥董偉兵孟繁博張俊杰
華北理工大學學報(自然科學版) 2012年1期
王帥,董偉兵,孟繁博,張俊杰
(1.河北聯合大學 化學工程學院,河北 唐山 063009;2.河北聯合大學 生命科學學院,河北 唐山 063009)
取代茚酮是重要的藥物合成前體,常用于藥物合成。茚酮結構廣泛存在于天然產物、藥物、農藥等生物活性分子中,也是有機發光、光致變色、染料等材料中的重要結構單元。近年來關于茚酮類化合物的報道較多[1-3]。利用光化學合成茚酮具有比以前的方法更簡潔、合成反應的中間步驟少、產率高等特點,且產物易分離、反應條件溫和[1]。本研究是對光化學合成6-甲基-1-茚酮方法的改進,極大地簡化了生產工藝,實現了綠色化學反應,具有較好的應用前景。
1 實驗部分
1.1 試劑和儀器
除非特殊說明,化學試劑一般為化學純,溶劑二甲苯為分析純。
Bruker EQUINOX 55型紅外光譜儀;Bruker 300 MHz核磁共振光譜儀;Thermo Finnigan Polaris Q氣質聯用儀(HP-5型毛細管柱);Hewlett Packard Model HP 6890 Series氣相色譜儀(HP-5型毛細管柱);VARIAN Cary-100 Bio紫外分光光度計;北京泰克儀器有限公司X-4型數字顯微熔點儀(熔點未校正)。
TLC板(自制)。
1.2 實驗方法
1.2.1 傅-克(Friedel-Crafts)酰基化反應
取對二甲苯19.0mL(150.0mmol)于100mL四口燒瓶中,加入3.0mL氯乙酰氯(37.1mmol),在氮氣保護下,加入5.50g(41.2mmol)無水三氯化鋁。在電磁攪拌和冰浴條件下,反應1小時。在室溫條件下攪拌2小時,可見大量鹽酸產生,可使濕潤試紙變紅。鹽酸應導入到氫氧化鈉溶液中吸收。必要時可以用石油醚稀釋。
反應的混合物,倒入100mL含有4mL濃鹽酸的冰水混合液中,放置到冰融化后,用100mL對二甲苯分兩次萃取,合并有機相并用水萃取至中性。利用硫酸鎂干燥過夜。砂芯漏斗中墊入少許硅膠,抽濾。TLC板檢測。
1.2.2 苯甲酸-(2,5-二甲基)-苯甲酰甲基酯的合成
稱取2,5-二甲基-苯甲酰甲基氯546mg(310mmol)溶解于50mL二甲苯溶液中,加入316mmol相應的羧酸和316mmol的三乙胺作為縛酸劑和500mg(310mmol)KI作為引發劑,在二甲苯中回流反應120min。反應的混合物經乙酸乙酯和水萃取,有機相用無水硫酸鈉干燥。抽濾,利用旋轉蒸發脫除有機溶劑后在石油醚中重結晶。TLC板檢測。
1.2.3 2,5-二甲基苯甲酰甲基氯的光解反應研究
用對二甲苯稀釋酰基化反應后的溶液,每加入10mL對二甲苯用TLC板監測一次,稀釋后的濃度以用TLC板監測時點的顏色深淺適中為宜。
取10mL溶液加入石英管中,通過改變溫度以及加入輔助試劑等在紫外燈下進行光化學反應,每二十分鐘監測一次,觀察是否有新點產生。
2 結果與討論
2.1 傅-克酰基化反應
試驗中我們將對二甲苯的用量過量,對二甲苯既作為反應原料,同時也是溶劑。反應產物經過處理和硅膠過濾后,溶液仍有些顏色,此時可以利用活性炭吸附脫色,以保證后續的光化學反應能夠有效的克服內濾光效應。
實驗產物經TLC板檢測,發現除對二甲苯原料外,新生成產物為單點。經過柱色譜分離產物,經過波譜表征,與文獻報道相同[1],證明產物 2,5-二甲基苯甲酰甲基氯生成。
圖1分別給出了2,5-二甲基苯甲酰甲基氯、苯甲酸-(2,5-二甲基)-苯甲酰甲基酯、6-甲基-1-茚酮在不同比例展開劑(乙酸乙酯:石油醚)中的遷移率曲線。由曲線1可以得出:應用乙酸乙酯:石油醚=1:20展開劑,可以有效地將傅-克酰基化反應中的產物2,5-二甲基苯甲酰甲基氯與弱極性的原料對二甲苯分離,達到純化2,5-二甲基苯甲酰甲基氯的目的。
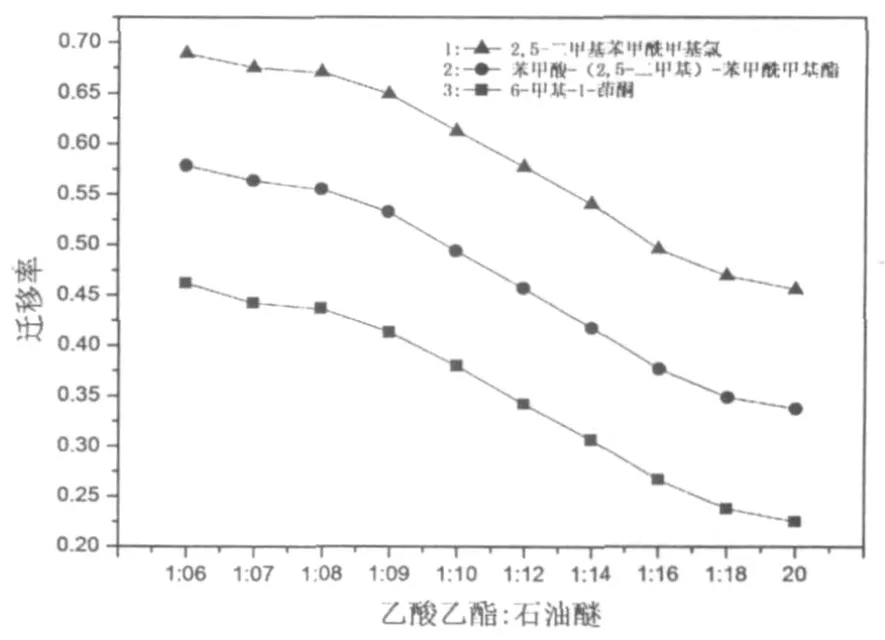
圖1 各級產物遷移率曲線
2.2 苯甲酸-(2,5-二甲基)-苯甲酰甲基酯的合成
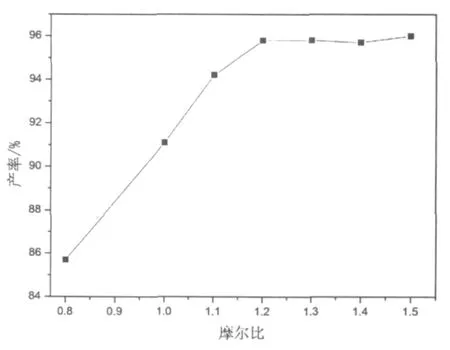
圖2 酯化反應三乙胺用量曲線
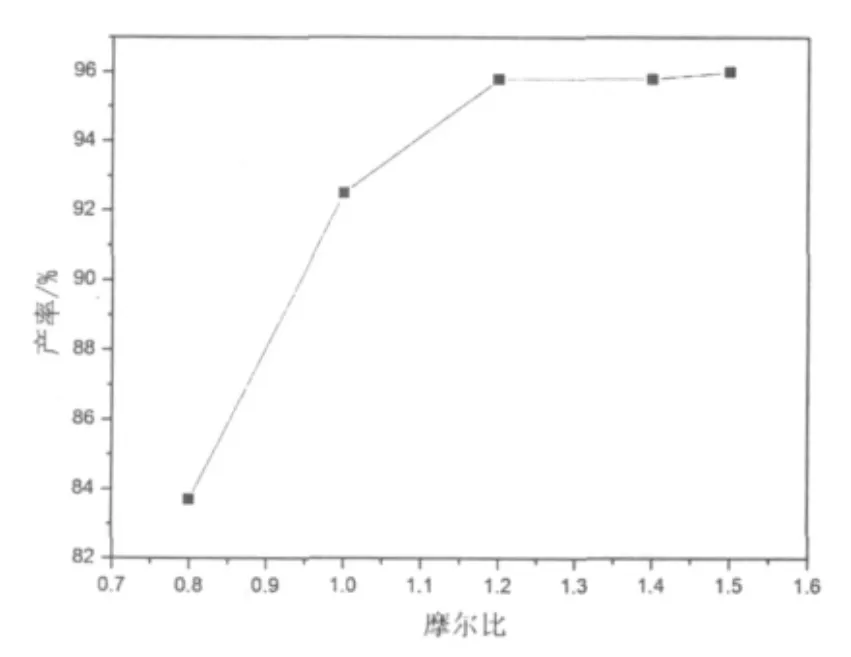
圖3 酯化反應苯甲酸用量曲線
2.2.1 三乙胺用量的影響
三乙胺能有效地催化2,5-二甲基苯甲酰甲基氯與苯甲酸的酯化反應,圖2給出了三乙胺用量對酯化反應產率的影響。由圖2可見,三乙胺的用量為2,5-二甲基苯甲酰甲基氯的1.2倍(摩爾比)時,酯化反應產率可以達到95%以上。
2.2.2 苯甲酸用量的影響
羧基是一個較氯原子更好的離去基團,文獻[1]曾用取代苯甲酰氯代烷酯化的方法得到相應的酯,通過光解進一步得到相應的取代茚酮。圖3給出了2,5-二甲基苯甲酰甲基氯與苯甲酸的酯化反應中苯甲酸的用量曲線。由圖3可見,苯甲酸的用量為2,5-二甲基苯甲酰甲基氯的1.2倍(摩爾比)時,酯化反應產率可以達到96%以上。
苯甲酸-(2,5-二甲基)-苯甲酰甲基酯的分離,可以采用石油醚中重結晶或柱色譜分離,圖1中曲線2和曲線3分別給出了苯甲酸-(2,5-二甲基)-苯甲酰甲基酯和6-甲基-1-茚酮在不同比例展開劑(乙酸乙酯:石油醚)中的遷移率(Rf),實驗結果表明:柱色譜分離純化6-甲基-1-茚酮可以乙酸乙酯:石油醚=1:20進行分離,去除2,5-二甲基苯甲酰甲基氯和苯甲酸-(2,5-二甲基)-苯甲酰甲基酯后,改用乙酸乙酯:石油醚=1:12的展開劑快速洗脫。收集的殘液可以進一步用于傅-克酰基化反應。苯甲酸-(2,5-二甲基)-苯甲酰甲基酯的表征數據與文獻報道相同[1]。
2.3 2,5-二甲基苯甲酰甲基氯的敏化光解反應
2.3.1 溫度對光解反應的影響
實驗研究了室溫和50℃條件下的2,5-二甲基苯甲酰甲基氯光解生成6-甲基-1-茚酮的現象。在室溫中,光解產物生成率較低,光解3小時產率約為5%;提高反應溫度對于光化學反應作用不大,由室溫升高到50℃時,光解3小時后產率提升至8%左右。
2.3.2 2,5-二甲基苯甲酰甲基氯光敏化現象的研究
考慮到苯甲酸-(2,5-二甲基)-苯甲酰甲基酯(量子效率0.23)比2,5-二甲基苯甲酰甲基氯(量子效率0.09)更容易光解的特點[1,4,5],本文選擇了在50℃條件下,加入三乙胺、苯甲酸和少量碘化鉀邊酯化邊光解的方法合成6-甲基-1-茚酮。實驗結果表明:在50℃條件下,光解2小時,6-甲基-1-茚酮約占50%,苯甲酸-(2,5-二甲基)-苯甲酰甲基酯有45%,2,5-二甲基苯甲酰甲基氯還剩大約5%。可見6-甲基-1-茚酮的合成速度得到明顯提高,速率大約提高了16倍,也表明了苯甲酸具有敏化光解反應的作用。本方法簡化了反應工藝和省略了酯化步驟,分離產物后的溶液可以進一步用于傅-克酰基化反應,節省了原料且減少了環境污染。
光解后的樣品經過稀氫氧化鈉水溶液萃取,水相和有機相分別保存。水相被進一步加熱除水,得到白色晶體,檢驗后可知為苯甲酸。有機相旋轉蒸發脫除溶劑,在石油醚中重結晶,得到相應的取代茚酮,或通過色譜柱分離。分離后的殘液,可以經過處理,再重新用于傅-克酰基化反應,實現了綠色化學反應。
2.3.3 光解反應及其機理的探討
根據分離的產物分析,我們推測光化學反應的機理如圖4所示。首先是在催化劑三乙胺的作用下,苯甲酸和2,5-二甲基苯甲酰甲基氯發生了酯化反應,該酯受到光照,吸收光子后,形成單線激發態,經過系間竄躍生產三重激發態。分子內發生1,5位氫轉移,產生了烯醇式形態的激發態,存在有順反兩種構型。顯然只有反式構型的形態有利于產生分子內環化,即形成茚酮。釋放出的苯甲酸進一步參與反應,如此反復。
余下的有機殘液進一步用于2,5-二甲基苯甲酰甲基氯的合成,這樣實現了綠色化學反應,無有機殘液排放,有利于環境保護,降低了生產成本。
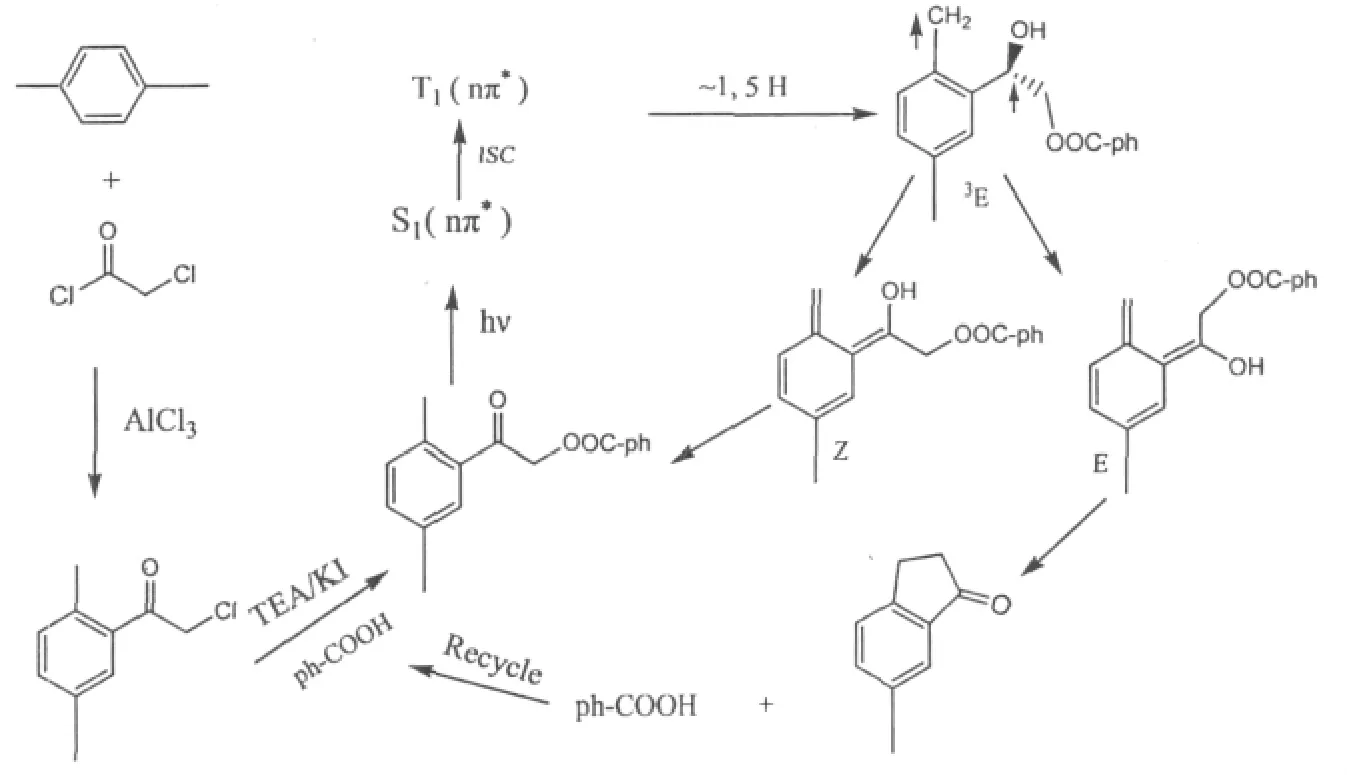
圖4 光敏化合成6-甲基-1-茚酮反應機理
3 結束語
本研究是對利用光誘導分子內氫轉移形成烯醇化反應進而有效合成取代茚酮的光化學方法的改進。方法充分利用了相對苯甲酰類保護劑而言,羧基比氯原子是更好離去基團的特點,通過溫和條件下的邊合成邊光解獲得了一個有效的合成取代茚酮的新方法。反應在50℃溫和條件下進行,極大地提高生成茚酮的速度,反應過程中只需加入少量的三乙胺和苯甲酸,由于生成茚酮后苯甲酸可以再和2,5-二甲基苯甲酰甲基氯反應,使反應循環進行,因此該方法簡單、光反應的產率高,產物易分離純化,而且具有綠色化學反應的特點,分離出產物后的殘液主要是對二甲苯,經過處理可重新用于傅-克酰基化反應。
[1]張俊杰,劉桂芳,李紅霞.一種光化學合成取代茚酮的有效方法[J].化學通報,2009,4:377-380.
[2]麻遠,殷巍,趙玉芬.1-茚滿酮合成方法研究進展[J].有機化學,2008,28(1):37-43.
[3]段義杰,劉建利,王翠玲.茚酮類化合物的研究進展[J].有機化學,2011,30(7):988-996.
[4]B.W.Rergmark,C.Barnes,J.Clark.et al.Photoenolization with α-Chloro Substituents[J].J.Org.Chem..1985,50(26):5612-5615.
[5]A.P.Pelliccioli,P.Klán,M.Zabadal et al.Photorelease of HCl from o-Methylphenacyl Chloride Proceeds through the Z-Xylylenol[J].J.Am.Chem.Soc.2001,123:7931-7932.
