配位化學中的鏡面對稱性破缺(續)*
——紀念配位化學創始人維爾納首次拆分八面體Co(Ⅲ)絡合物100周年
2012-09-25 03:02:22章慧林麗榕
大學化學 2012年1期
章慧 林麗榕
(廈門大學化學化工學院化學系 福建廈門 361005)
2 配位化學中的MSB實例及其機理和應用研
2.1 經典絡合物cis-[CoBr(NH3)(en)2]Br2的絕對不對稱合成
盡管Asakura等早在1990年就偶然發現在cis-[CoBr(NH3)(en)2]Br2的制備過程中存在著MSB現象(圖3)[7,63],但直到1995年以ASS獲得手性cis-[CoBr(NH3)(en)2]Br2的結果才被正式報道。1995年起,Asakura等基于該絡合物絕對不對稱合成的一系列研究認為[7,30,63-67],其反應機理是由于手性鈷絡合物晶體誘導自身產生的不對稱自催化過程。自2003年至今,我們在Asakura的實驗基礎上[7],經過反復摸索和優化實驗條件,重復了Asakura的實驗結果[9-10],分別獲得了Δ-、Λ-和消旋體3種產物的單晶結構[10],對手性產物進行了溶液和固體CD光譜表征,同時大膽提出了自己假設的“電子轉移催化-結晶誘導”機理[9]。我們還將cis-[CoBr(NH3)(en)2]Br2的制備和表征改造成了一個綜合化學實驗[8],從2004年起在廈門大學化學系碩士生中實施。從1993年至2008年,我系先后共有100名學生和教師進行了376次重復實驗,其中獲得Λ構型產物164次、外消旋體141次、Δ構型產物71次;顯然,獲得Λ構型產物的概率大約是獲得Δ構型產物的2倍,獲得消旋體與獲得Λ構型產物的次數基本持平。由此創下了這一ASS實驗在國際上統計數據的新紀錄。
十分有趣的是,自2006年起,Coogan等也試圖重復Asakura的ASS實驗,但是始終未能得到手性產物[68-69]。起先也未獲得非手性合成的外消旋化合物cis-[CoBr(NH3)(en)2]Br2的理想單晶,因此只能對其微晶進行了XRD表征,并擬合了其晶體結構,空間群為P21/n[68]。之后,該同質多晶型(Ⅲ-相)外消旋cis-[CoBr(NH3)(en)2]Br2進一步被單晶結構分析證明[10,68]。

圖14 惰性絡合物cis-[CoBr(NH3)(en)2]Br2的絕對不對稱合成
經過數百次重復實驗的仔細觀察、實驗數據分析和認真思考后,我們對Asakura提出的“不對稱自催化”機理產生了懷疑,理由如下[9]:① 已知在“結晶誘導的不對稱轉化”反應中,外消旋混合物中的一對對映體必須具備在某種條件下容易發生消旋體轉換(racemic switch)的性質;但是,Co(Ⅲ)絡合物是取代動力學上惰性的,不論是Δ-Co(Ⅲ)絡合物轉化為Λ-Co(Ⅲ)絡合物,或是其相反的過程都難于實現;即便有某一對映體的晶種(例如Δ-Co(Ⅲ)或Λ-Co(Ⅲ)絡合物單晶)的誘導,也不太可能從另一種對映體很容易地轉化過來。② 磁化率測定證明在起始反應物(非手性三核鈷絡合物)中,存在著一個Co(Ⅱ)中心(如圖14所示),當三核鈷絡合物中的配位鍵在溴化銨和水的作用下發生斷裂形成手性單核Co(Ⅲ)絡合物時,體系中客觀存在的占鈷總量為1/3的單核Co(Ⅱ)物種究竟起著什么樣的作用(已知Co(Ⅱ)絡合物是取代活性的)?Asakura等似乎將它們忽略了。③ Co(Ⅱ)/Co(Ⅲ)胺合絡合物體系的內界電子轉移(ET)反應速率相當快,而且經設計的ET催化不對稱合成早已成功地應用于合成[Co(en)3]3+的單一手性對映體等類似體系[49]。④ 在合成中,反應產物的產率和純度會隨著反應的粗產物(混合物)在冰箱中放置時間的增加而獲得改善,值得注意的是,此時Co(Ⅱ)物種仍舊留在反應體系中。
顯然,上述所觀察的實驗現象與Asakura等提出的“不對稱自催化機理”相悖,反應過程中可能存在促使惰性手性Co(Ⅲ)絡合物外消旋化的因素。基于此,我們于2006年提出了電子轉移催化-結晶誘導的假設機理[9]:“Δ-或Λ-cis-[CoBr(NH3)(en)2]Br2單一對映體的形成是以Co(Ⅱ)物種為催化劑,通過Co(Ⅱ)/(Ⅲ)電子轉移促進Co(Ⅲ)絡合物外消旋轉化,同時結合手性晶體的誘導成核的絕對不對稱合成機理”。我們相信,該反應的真正機理之謎一旦能被解開,將對配位化學中MSB成因的基礎研究和類似惰性化合物的絕對不對稱合成設計有著非同尋常的意義。對于我們提出的新機理,還需要通過精心設計各種相關動力學實驗以獲取更多的實驗數據來進行論證。
為了進一步應用ASS獲得的惟手性金屬中心鈷絡合物,我們還嘗試將手性cis-[CoBr(NH3)(en)2]Br2作為手性源,用以誘導卟啉的超分子手性聚集與組裝(圖15),使“無中生有”獲取的手性因素得到成功傳遞,繼而實現了手性記憶和手性放大[70]。

圖15 “無中生有”的手性-手性Co(Ⅲ)絡陽離子誘導卟啉的超分子手性聚集與組裝
2.2 含聯吡啶和鄰菲洛啉的Ru(Ⅱ)絡合物
如本文1.4中所述,由于惰性絡合物在溶液中不易發生外消旋轉化,即便發生鏡面對稱性破缺,其過程一般為自發拆分,而不是絕對不對稱合成。例如,已知[Ru(bpy)3](PF6)2有4種同質多晶型(表2)[71-74],α和β型是外消旋化合物,γ和δ型是手性化合物;其中,γ型晶體由酒石酸銻鉀拆分獲取,而δ構型的準同質多晶型單晶則是從丙酮溶液中經自發拆分而得。

表2 [Ru(bpy)3](PF6)2 4種同質多晶型的晶體結構參數
*該構型為含丙酮溶劑的準同質多晶型δ-[Ru(bpy)3](PF6)2·1.5C3H6O。
2006年,黃偉等在合成[Ru(phen)(bpy)2](PF6)2的過程中發現了伴隨著共生同質多晶的MSB現象(表3)[75]。雖然在涉及這類惰性Ru(Ⅱ)絡合物MSB的兩篇文獻中[74-75],都沒有對大宗產物作出CD光譜表征,無法判斷該MSB過程究竟是自發拆分還是絕對不對稱合成,但是在這兩個實例中,我們都可以看到溶劑分子在形成Congl.時所扮演的重要角色。

表3 [Ru(phen)(bpy)2](PF6)2兩種同質多晶型的晶體結構參數[75]
*該構型為含結晶水的準同質多晶型α-[Ru(phen)(bpy)2](PF6)2·C2H5OH·H2O。
2.3 手性格氏試劑、鈀催化劑和鋅試劑——MSB在不對稱合成中的應用
迄今,已有化學家利用已獲取的MSB實驗結果,設法將所得手性化合物所含有的手性信息固定下來,并巧妙地應用于不對稱合成過程。以下簡要介紹3個典型實例。
圖16所示的具有C2對稱性的手性雙膦配體(biphos)[76]結合了分子骨架軸手性和磷原子手性特征。理論上講,該配體共有6個光學異構體存在,相當于3對對映異構體。由于biphos在溶液中是一種動力學活性(具有不穩定的軸手性和磷原子手性)的化合物,這些光學異構體之間會迅速達到平衡而形成消旋體系。在固態,這種立體構型的不穩定性得以克服。Balavoine等利用結晶誘導的不對稱轉化反應,使biphos的某一單一光學異構體在溶液中析出得到的大單晶(質量約為50~100mg)在低溫(-78℃)下與[PdCl2(CH3CN)2]反應,手性選擇性地定量合成了高對映純度的、手性構型穩定的絡合物S-[R,R]-(+)-[PdCl2(biphos)]和R-[S,S]-(-)-[PdCl2(biphos)](前綴首字母表示骨架軸手性,方括號中的符號表示配位磷原子的手性),接著將該手性鈀絡合物應用于催化不對稱烷基取代反應,獲得了高產率和較好的ee值。在優化條件下,當采用R-[S,S]-(-)-[PdCl2(biphos)]為手性催化劑,得到R-構型的烷基化產物;而當采用S-[R,R]-(+)-[PdCl2(biphos)]時,得到S-構型的產物。最終實現了絕對不對稱合成→低溫下的配位反應(手性固定和傳遞)→催化不對稱合成(生成新手性碳中心的手性信息轉移和增殖),堪稱手性“無中生有”,最后得到不對稱催化增殖的經典范例。

圖16 將“結晶誘導的不對稱轉化”所得大單晶biphos應用于催化不對稱烷基取代反應
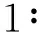
H?kansson等還利用絕對不對稱合成對映純有機鋅試劑晶體,并將其在低溫下研磨溶解于甲醇中,迅速與NCS(N-chlorosuccinimide)反應,以高產率和高對映選擇性得到手性氯茚[80](圖18)。

圖17 惟手性金屬中心的格氏試劑的ASS及其誘導的化學計量不對稱加成反應

圖18 以ASS獲取手性鋅試劑及其誘導的化學計量不對稱有機反應
以上3個例子中都應用了結晶誘導的不對稱轉化策略來實現化合物手性的“無中生有”(還可通過事先加入手性晶種來調控MSB產物的手性構型),并根據相應體系分別精巧設計了不對稱催化和化學計量的手性誘導來實現有機不對稱合成的目標(雖然在手性格氏試劑的例子中最終產物的ee值還比較低),這說明奇妙的MSB現象完全可以被加以利用,類似的方法可望再進一步充實、完善,從而發展成不對稱合成的一般方法。但是由于在這3個研究工作中都實施了低溫、Schlenk技術或其他較苛刻的反應條件,因此可能使之成為較難被超越且被進一步推廣應用的個例。
2.4 活性稀土絡合物
7配位手性稀土絡合物[Ln(H2O)(dmb)3](Ln=La,Pr,Sm,Er,Eu,Nd,Tb,Dy;Hdmb=dibenzoylmethane)也可由結晶誘導的不對稱轉化獲得[33-34,81],H?kansson等的實驗結果表明,所得單一手性產物的產率為60%~94%,對映體過量百分率分布在97%~100%ee之間,可稱之為ASS反應[33]。H?kansson等發現:實驗中總是獲得Δ-[Ln(H2O)(dmb)3],因此推測他們的實驗室里可能存在某種痕量的手性雜質影響著結晶過程,雖然十分小心地操作以避免Δ-[Sm(H2O)(dmb)3]晶種的“污染”,但它們的存在仍難以被排除。他們采用Δ-[Co(acac)3]晶種進行手性誘導后才得到Λ-[Sm(H2O)(dmb)3];而一旦獲得了Λ-構型的單晶并將其作為晶種,則控制大宗結晶產物為Λ-構型變得輕而易舉。十分有趣的是,試圖從這類體系中獲得外消旋產物比獲得單一對映體產物的難度更大。而我們的研究結果則表明[81],不需加入晶種,而是從其中任意一種單一手性的樣品出發,通過對其進行反復多次重結晶就可以得到與之相反手性的7配位稀土Eu絡合物(圖19~21),由此說明該化合物的ASS機理,是由體系中隨機產生的第一顆手性晶種的誘導來實現的。與手性格氏試劑的ASS類似,這類化合物因結晶獲得的金屬中心手性在溶液中即刻消失,所以探討其在室溫下溶液中的進一步應用沒有實際意義。

圖19 Δ和Λ-[Eu(H2O)(dmb)3]的外消旋體不對稱轉化

圖20 Δ-[Eu(H2O)(dmb)3](左)和Λ-[Eu(H2O)(dmb)3](右)的晶體結構
為了驗證配位水分子在晶體堆砌中是否能通過氫鍵進行手性傳遞,我們對7配位手性稀土絡合物Δ-Sm、Δ-Eu和Δ-Tb的手性晶體進行了加熱抽真空脫水成黃色粉末以及吸水后又重新轉化為手性晶體的觀察[81],發現脫水后的絡合物基本失去了固體CD信號,而吸水后重得手性晶體的CD信號又恢復如初,由此可認為配位水分子在晶體堆砌的過程中確實起著通過氫鍵鏈接傳遞手性的作用[33,81],進一步證實了晶體結構分析的結果。

圖21 由Δ-[Eu(H2O)(dmb)3]大宗手性晶體出發經10次重結晶所得固體CD譜KCl壓片,0.1%
H?kansson等在1999年還利用自發拆分獲得了8配位畸變六角雙錐型手性稀土絡合物Δ/Λ-trans-[SmI2(dme)3][45],其中的兩個I原子呈反式排列,可根據3個dme配體的螺旋排列方式并結合其Flack參數將其絕對構型指認為Δ或Λ。除了在f-f 躍遷區可能有微弱的CD信號外,手性trans-[SmI2(dme)3]缺乏合適的紫外可見CD生色團,因此無法對所獲得單晶進行固體CD表征,只能通過晶體結構和偏光顯微鏡來鑒別對映體。
2009年,H?kansson等在9配位稀土絡合物Na5[Ln(oda)3](H2O)6(BF4)2(Ln=Dy、Er和Pr,H2oda=一縮二乙二醇酸)的合成中也發現了MSB現象(表4)[55]。該文未能成功地對這3個Congl.化合物的大宗產物進行固體CD光譜表征,筆者推測,這可能是由于該系列化合物缺乏合適的CD生色團或該文所實施固體CD測試方法不當的緣故。但H?kansson等通過控制結晶可使Na5[Er(oda)3](H2O)6(BF4)2溶液中所有溶質形成一顆大單晶,對取自該單晶不同部分的4個碎片分別進行的晶體結構分析表明,它們均有相同的絕對構型,基于Na5[Ln(oda)3](H2O)6(BF4)2在溶液中是活性的,故將該合成反應定義為ASS。十分有趣的是,當體系中不存在NaBF4,或者用NH4SCN代替NaBF4時,所形成的Na3[Gd(oda)3](H2O)6和Na3NH4[Pr-(oda)3](SCN)(H2O)4為外消旋化合物。此實驗事實說明,與表1所示的過渡金屬絡合物類似,稀土絡合物所含的抗衡陰(陽離子)在形成Congl.時同樣起重要作用,但其成因尚不清楚。

表4 9配位稀土絡合物[Ln(oda)3]3-的晶體結構參數


圖22 Na5[Dy(oda)3](H2O)6(BF4)2的晶體結構

圖23 Na5[Dy(oda)3](H2O)6(BF4)2晶體中呈現的各種手性子結構
2.5 含三腳架型配體的第一過渡系金屬絡合物
迄今為止,MSB現象被認為是不可預測的,但根據文獻報道,在類似Na5[Ln(oda)3](H2O)6(BF4)2中發現的準三方對稱性結構發生MSB的概率顯然較大,其中以三腳架型配體形成的第一過渡系絡合物較常見[82-87]。


圖24 三角架式tren配體的4種配位螯環手性構象

圖25 Das等提出的cis-[Ni(NCS)2(tren)]的一對“對映體”

圖26 (λλδ)-cis-[Ni(NCS)2(tren)](左)和(δδλ)-cis-[Ni(NCS)2(tren)] (右)的配位環境

圖27 半面晶外觀不同的一對Co(Ⅱ)絡合物的對映體(A)和(B)及其固體CD光譜和絕對構型關聯
Matsumoto等自行設計合成了三腳架型六齒氮配體(圖27),并對自發拆分所形成風扇形M(Ⅱ)絡合物(M=Co、Mn、Fe、Ni和Zn)的晶體結構及其固體CD光譜進行了關聯[85-87],其CD光譜特征反映了金屬中心手性(構型效應)的貢獻以及配體的螺旋手性構象(δδδ或λλλ)對d-d躍遷生色團或配體π-π*生色團的手性微擾。
與Pasteur當年的工作非常相似,Matsumoto等利用半面晶外觀的不同(圖27),手工分離出一對以Congl.形式結晶的消旋Co(Ⅱ)絡合物的對映體。在結構關聯研究中,將同一顆Co(Ⅱ)絡合物單晶(Sohncke空間群P212121)既用于晶體結構分析也用于固體(KBr壓片)CD光譜測定,即進行晶體形狀、絕對構型(C或A,分別表示順時針或反時針螺旋方向)和CD光譜關聯。其他經自發拆分的同構Mn(Ⅱ)、Fe(Ⅱ)、Ni(Ⅱ)和Zn(Ⅱ)絡合物的絕對構型可以與約320nm處的Cotton效應符號進行關聯。與相應的Co(Ⅱ)絡合物類似,其他M(Ⅱ)絡合物所形成外消旋混合物中的一對對映體也具有不同的半面晶觀。
2.6 Re(Ⅴ)絡合物
在合成如圖28所示的手性Re(Ⅴ)絡合物cis-[ReOCl2{OCMe2CMe2OP(OCMe2CMe2O)}(py)](Sohncke空間群P212121)過程中也觀察到MSB現象[88-89]。但與上面介紹的活性絡合物不同的是,其手性產物在溶液中的構型相對穩定,可以測得單晶的溶液CD光譜。已知該絡合物的外消旋轉化經歷了一個可溶的非手性trans-中間體的過程。具有一種對映體大過量的手性產物的產率接近70%,利用純對映體的固體粉末進行播晶亦可誘導單一手性產物的形成。

圖28 cis-Re(Ⅴ)絡合物的結晶誘導不對稱轉化過程

圖29 手性cis-Re(Ⅴ)絡合物的ee值分布
圖29a表明,當cis-[ReOCl2{OCMe2CMe2OP(OCMe2CMe2O)}py]的不對稱轉化在沸騰的甲苯溶液中加熱和劇烈的攪拌條件下進行時,呈現兩種對映體產物的隨機分布;既不煮沸也不攪拌的加熱條件下則主要產生消旋體,即反應體系處于外消旋平衡中(圖29b);當采用播晶方法時,不對稱轉化定向進行并呈現高ee值(圖29c和d)。
2.7 阻轉異構絡合物
相對于易發生MSB的阻轉異構有機化合物[90],對阻轉異構金屬絡合物MSB現象的報道不多,下面簡要介紹其中兩例[91-92]。
Faller等采用W(CO)6、1-氯-2-丁烯(crotyl chloride)與非手性雙膦配體diphos反應制得syn-[W(CO)(η3-crotyl)(diphos)Cl],晶體結構分析和變溫NMR表明該合成反應呈現MSB現象,反應主產物為圖30所示的(S)-(A)-[W(CO)(η3-crotyl)(diphos)Cl](Sohncke空間群P212121),屬于一種ASS。通過不同物種之間的差向異構化,“無中生有”地產生了所得手性絡合物配體巴豆基(η3-crotyl)上的手性碳和苯基磷的阻轉異構螺旋手性[91]。Forniés等也發現金屬有機絡合物[{Li(thf)3}2(μ-Cl)]2]-[CrⅡ(C6Cl5)4](圖31)結晶時析出的單晶為Congl.(Sohncke空間群I4)[92],其晶體結構顯示為一種罕見的準平面四方形螺旋槳狀手性構象,即4個氯苯在中心金屬周圍呈M螺旋排列,但是,Forniés等并沒有通過其他表征手段來證明該合成反應究竟是自發拆分還是ASS。

圖30 準八面體W絡合物的非對映異構體構型轉換
2.8 雙核絡合物和金屬籠絡合物的MSB現象
多核絡合物的MSB研究以配位聚合物居多,我國科學家在這一方面進行了高水平的研究[11-27],Pérez-García和Amabilino對此曾經作出很好的綜述[27]。相比之下,在雙核或寡聚金屬籠絡合物中發現MSB的實例甚少。以下著重介紹幾例中國學者的工作。
段春迎等利用自行設計合成的配體L1和L2(圖32)分別制備了兩個雙核銀絡合物[93],在[Ag2(L1)2](NO3)2·CH3OH的產物中發現一對對映體,其空間群分別為P3121(M構型)和P3221(P構型),對同一批次結晶中的20顆單晶進行的固體CD表征發現,該合成反應的ee值為零,為自發拆分;而在[Ag2(L2)2](ClO4)2的合成中,亦獲得空間群分別為P3121(M構型)和P3221(P構型)的一對對映體。對50個批次重結晶(每批次取20顆單晶分析)的固體CD光譜研究表明[Ag2(L2)2](ClO4)2的制備反應主要為ee值過量的ASS,但手性產物的構型是隨機分布的。王梅等利用圖32所示的配體L3,分別制備了同構的兩對手性雙核鋯和雙核鉿絡合物[94],其空間群均為P212121。對雙核鋯的一對對映體單晶進行了固體CD光譜表征,大宗產物的固體CD分析表明同一批次結晶的ee值為0,為自發拆分過程。值得一提的是,基于L3制備的手性雙核鋯(鉿)絡合物的的手性立體化學結構特征,是該配體中的N原子在配位后成為手性中心,并通過結晶過程的自發拆分得到了較為罕見的兩對對映體。

圖31 金屬有機絡合物[{Li(thf)3}2(μ-Cl)]2[CrⅡ(C6Cl5)4](Sohncke空間群I4)中絡陰離子的結構
近年來,采用C2、C3或C4對稱性的多齒配體設計合成四面體、八面體或其他幾何構型金屬籠絡合物的研究已成為超分子化學的研究熱點之一[61,95-99],而手性籠的設計與合成也是其中的重要部分。迄今,手性籠絡合物主要靠手性配體的立體選擇性合成獲得[98],被拆分的實例很少,而能夠被自發拆分的則更加罕見。Raymond等發現利用形成四面體籠狀M(Ⅲ)(M=Ga、Al、Fe)絡合物的三維框架結構,并輔以籠內陽離子的支撐,可以有效地“鎖住”位于四面體角頂活性金屬中心(6配位八面體構型)的同手性ΔΔΔΔ-或ΛΛΛΛ-構型,抵御其外消旋化,并以尼古丁衍生物為拆分劑實現了它們的拆分[61]。然而,由于多數角頂金屬的取代活性以及同手性Δ和Λ兩種構型的籠絡合物在熱力學上具有相同穩定性,在溶液中形成兩種相反手性構型的金屬籠的概率是均等的,采用非手性配體合成所得固體產物通常是外消旋化合物,但不排除其在合成和結晶過程中出現MSB現象。例如,何成等的研究表明[95],所設計合成的四面體籠狀M(Ⅱ)(M=Zn和Cu)絡合物在結晶時可獲得手性單晶,其所屬空間群為P3。我們在對其晶胞中的3個籠分子進行結構分析時發現,這3個絡合物并不具有相同的絕對構型,如圖33所示,這種情況與本文1.2中所述的外消旋體的第3種形態(兩種對映異構體以非等量的形式存在于晶格中)的假外消旋體十分相似。因此建議:若某化合物的單晶被解析為手性空間群或Sohncke空間群,當晶胞中含有多個分子時,必須逐個考察它們的絕對構型,以正確判斷該化合物的結晶行為究竟屬于外消旋體的何種形態。

圖32 用于合成雙核絡合物的非手性多齒配體

圖33 四面體籠絡合物的晶體結構——兩種對映異構體以非等量的形式存在于晶胞中
3 結束語
立體化學的發展與手性化合物的研究密切相關。在三維空間上了解分子的結構和性能,已成為化學、材料和生命科學深入研究的課題,當今在相關前沿領域的任何重要發現和突破,離開了立體化學的理解、闡述和指導都是難以想像的[100],配位化學的發展也不例外。自維爾納對八面體絡合物進行首次拆分,并奠定了配位立體化學基礎以來,鏡面對稱性破缺現象研究已經成為現代手性配位立體化學的一個重要組成部分。由此所進行的相關結構和機理的深入探究,或許在不遠的將來可用于MSB現象的預測和調控,使ASS能夠真正成為獲取對映純手性化合物的一個簡便方法。
參 考 文 獻
[1] 章慧.配位化學——原理與應用.北京:化學工業出版社,2009
[2] Meggers E.EurJInorgChem,2011(19):2911
[3] Kauffman G B.CoordChemRev,1974,12(2):105
[4] Kauffman G B,Bernal I.JChemEdu,1989,66(4):293
[5] Bernal I,Kauffman G B.JChemEdu,1987,64(7):604
[6] Bernal I,Kauffman G B.StructChem,1993,4(2):131
[7] Asakura K,Kobayashi K,Mizusawa Y,etal.PhysicaD,1995,84(1-2):72
[8] 方雪明,章慧,陳雷奇,等.大學化學,2006,21(2):48
[9] 章慧,王憲營,陳雷奇,等.物理化學學報,2006,22(5):609
[10] 丁冬冬.手性金屬配合物的不對稱合成和氧化還原手性開關應用初探.廈門大學碩士學位論文,2011
[11] Han L,Hong M C,Wang R H,etal.ChemCommun,2003,2580
[12] Gao E Q,Yue Y F,Bai S Q,etal.JAmChemSoc,2004,126(5):1419
[13] Qu Z R,Zhao H,Wang Y P,etal.ChemEurJ,2004,10(1):53
[14] Wu S T,Wu Y R,Kang Q Q,etal.AngewChemIntEd,2007,46(44):8475
[15] Li X Z,Li M,Li Z,etal.AngewChemIntEd,2008,47(34):6371
[16] Lan Y Q,Li S L,Su Z M,etal.ChemCommun,2008(1):58
[17] Hou S Z,Cao D K,Li Y Z,etal.InorgChem,2008,47(22):10211
[18] Zhang S Y,Yang S G,Lan J B,etal.ChemCommun,2008(46):6170
[19] Zhang J,Chen S M,Wu T,etal.JAmChemSoc,2008,130(39):12882
[20] Chen H F,Guo G C,Wang M S,etal.DaltonTrans,2009,38(46):10166
[21] Liu Y,Xuan W M,Zhang H,etal.InorgChem,2009,48(21):10018
[22] Tong X L,Hu T L,Zhao J P,etal.ChemCommun,2010(46):8543
[23] Su Z,Chen M S,Fan J A,etal.CrystEngComm,2010,12(7):2040
[24] Liu L,Huang S P,Yang G D,etal.CrystGrowthDes,2010,10(2):930
[25] Zheng X D,Lu T B.CrystEngComm,2010,12(2):324
[26] Liu J L,Bao X,Leng J D,etal.CrystEngComm,2011,11(6):2398
[27] Pérez-García L,Amabilino D B.ChemSocRev,2007,36(6):941
[28] 卜顯和,章慧.手性自發拆分的預測和調控∥10000科學難題·化學卷.北京:科學出版社,2009:29-31
[29] Gourlay M D,Kendrick J,Leusen F J J.CrystalGrowth&Design,2007,7(1):56
[30] Kondepudi D K,Asakura K.AccChemRes,2001,34(12):946
[31] Kondepudi D K,Kaufman R J,Singh N.Science,1990,250(4983):975
[32] Asakura K,Nagasaka Y,Osanai S,etal.JPhysChemB,2005,109(4):1586
[33] Lennartson A,Vestergren M,H?kansson M.ChemEurJ,2005,11(6):1757
[34] Zhou N,Wan S G,Zhao J,etal.ScienceinChinaSeriesB:Chemistry,2009,52(11):1851
[35] Lennartson A,H?kansson M.AngewChemIntEd,2009,48(32):5869
[36] 丁奎嶺,王洋,吳養潔,等.有機化學,1996,16(1):1
[37] 李越蘭,關燁第.大學化學,1997,12(1):19
[38] Flack H D.HelvChimActa,2003,86(4):905
[39] Müller U.Inorganic Structural Chemistry.2nd ed.West Sussex:John Wiley & Sons,Ltd,2007
[40] 伊萊爾E L,威倫S H,多伊爾M P.基礎有機立體化學.鄧并主譯.北京:科學出版社,2005
[41] Koshima H,Ding K L,Chisaka Y,etal.JAmChemSoc,1996,118(48):12059
[42] 俞蕓,林麗榕,楊開冰,等.有機化學,2006,26(7):933
[43] Bernal I,Cetrullo J,Jackson W G.InorgChem,1993,32(19):4098
[44] Yamanari K,Hidaka J,Shimura Y.BullChemSocJpn,1973,46(12):3724
[45] H?kansson M,Vestergren M,Gustafsson B,etal.AngewChemIntEd,1999,38(15):2199
[46] Lennartson A,Olsson S,Sundberg J,etal.AngewChemIntEd,2009,48(17):3137
[47] Lennartson A,Olsson S,Sundberg J,etal.InorgChimActa,2010,363(1):257
[48] Vestergren M,Eriksson J,H?kansson M.ChemEurJ,2003,9(19):4678
[49] 王尊本.綜合化學實驗.第2版.北京:科學出版社,2007
[50] Hamelin O,Pécaut J,Fontecave M.ChemEurJ,2004,10(10):2548
[51] Alvarez S,Alemanyb P,Avnir D.ChemSocRev,2005,34:313
[52] Bosnich B,Sullivan E A.InorgChem,1975,14(11):2768
[53] Mason S F.Molecular Optical Activity and the Chiral Discriminations.Cambridge:Cambridge University Press,1982
[54] 金斗滿,朱文祥.配位化學研究方法.北京:科學出版社,1996
[55] Lennartson A,H?kansson M.CrystEngComm,2009,11(9):1979
[56] Faller J W,Parr J,Lavoie A R.NewJChem,2003,27(6):899
[57] Ziegler M,von Zelewsky A.CoordChemRev,1998,177(1):257
[58] Richardson F S.ChemRev,1979,79(1):17
[59] Bosnich B,Harrowfield J M.JAmChemSoc,1972,94(10):3425
[60] Telfer S G,Tajima N,Kuroda R.JAmChemSoc,2004,126(5):1408
[61] Davis A V,Fiedler D,Ziegler M,etal.JAmChemSoc,2007,129(49):15354
[62] Lama M,Mamula O,Kottas G S,etal.InorgChem,2008,47(18):8000
[63] Asakura K,Osanai S,Kondepudi D K.Chirality,2001,13(8):435
[64] Asakura K,Kondepudi D K,Martin R.Chirality,1998,10(4):343
[65] Asakura K,Inoue K,Osanai S,etal.JCoordChem,1998,46(2):159
[66] Asakura,K,Ikumo A,Kurihara K,etal.JPhysChemA,2000,104(12):2689
[67] Burch K W,Burch M A.Chiral Analysis.Amsterdam:Elsevier,2006
[68] Guo F,Casadesus M,Cheung E Y,etal.ChemComn,2006,22(17):1854
[69] Casadesus M,Coogan M P,Davies E,etal.InorgChimActa,2008,361(1):63
[70] Wang J,Ding D D,Zeng L X,etal.NewJChem,2010,34(7):1394
[71] Bine M,Burgi H B,Ludi A,etal.JAmChemSoc,1992,114(13):5197
[72] Rillema D P,Jones D S,Woods C,etal.InorgChem,1992,31(13):2935
[73] Breu J,Domel H,Stoll A.EurJInorgChem,2000(11):2401
[74] Breu J,Seidl W,Huttner D,etal.ChemEurJ,2002,8(19):4454
[75] Huang W,Ogawa T.Polyhedron,2006(25):1379
[76] Tissot O,Gouygou M,Dallemer F,etal.AngewChemIntEd,2001,40(6):1076
[77] Vestergren M,Eriksson J,H?kansson M.ChemEurJ,2003,9(19):4678
[78] Vestergren M,Gustafsson B,Davidsson ?,etal.AngewChemIntEd,2000,39(19):3435
[79] Vestergren M,Eriksson J,H?kansson M.JOrganometChem,2003,681(1):215
[80] Lennartson A,Olsson S,Sundberg J,etal.AngewChemIntEd,2009,48(17):3137
[81] 宣為民.β-二酮配合物的拆分、不對稱合成及其固體圓二色光譜研究.廈門大學碩士學位論文,2008
[82] Rao A S,Pal A,Ghosh R,etal.InorgChem,2009,48(5):1802
[83] Richard M H,Rob S.InorgChem,2009,48(21):10476
[84] 劉成勇,顏建新,林以璣,等.物理化學學報,2011,DOI:10.3866/PKU.WHXB201112091
[85] Ikuko K,Naohide M,Masaaki K.InorgChem,2000,39(15):3350
[86] Nagasato S,Katsuki I,Motoda Y,etal.InorgChem,2001,40(11):2534
[87] Katsuki I,Motoda Y,Sunatsuki Y,etal.JAmChemSoc,2002,124(4):629
[90] Ding L,Lin L R,Liu C Y,etal.NewJChem,2011,35(9):1781
[91] Faller J W,Fontaine P P.JOrganometalChem,2006,691(22):4667
[92] Alonso P J,Forniés J,García-Monforte M A,etal.ChemEurJ,2002,8(17):4056
[93] Sun Q J,Bai T,He G J,etal.ChemCommun,2006(26):2777
[94] Hu M G,Wang M,Zhu H J,etal.DaltonTrans,2010,39(18):4440
[95] He C,Wang L Y,Wang Z M,etal.DaltonTrans,2002(2):134
[96] Bryan G S,Tiedemann E F,Raymond K N.TopCurrChem,2006,265:147
[97] Mal P,Schultz D,Beyeh K,etal.AngewChemIntEd,2008,47(43):8297
[98] Liu T,Liu X W M,Cui Y.AngewChemIntEd,2010,49(24):4121
[99] Meng W J,Breiner B,Rissanen K,etal.AngewChemIntEd,2011,123(15):3541
[100] 尤田耙,林國強.不對稱合成.北京:科學出版社,2006
