提鋰吸附劑的合成與吸附過程研究
2012-10-17 02:51:30解利昕陳小棉
無機鹽工業 2012年12期
解利昕 ,趙 煜 ,陳小棉 ,解 奧
(1.天津大學化工學院,天津市膜科學與海水淡化技術重點實驗室,天津 300072;2.天津大學理學院)
近年來,隨著鋰電池需求量迅速提升,鋰資源開發成為研究熱點[1-3]。但是,鋰資源分布極不平衡,陸地上鋰資源僅是海洋中的萬分之一[4-6]。而海水中鋰元素濃度極低(質量濃度僅為 0.17 mg/L)[7],如何在極低濃度下吸附鋰元素成為從海水中提取鋰元素所面臨的主要問題[1]。因此,海水提鋰已經成為世界各國科學家與工程技術人員努力研究的重要課題。其中開發出能夠在低濃度下具有吸附效果的吸附劑成為海水提鋰的關鍵。日本的Ramesh Chitrakar等、韓國的Kang-Sup Chung等以及中國的袁俊生、王祿等對提鋰吸附劑的制備與應用做了基礎性研究[6,8-10]。在已知的諸多吸附劑中,尖晶石型鋰錳氧化物對鋰離子的吸附作用得到了較多關注。尖晶石型鋰錳氧化物是一系列化合物,包括Li1.33Mn1.67O4、LiMn2O4、Li1.6Mn1.6O4等,其對鋰離子吸附原理[11]主要分為氧化還原法和離子交換法兩種類型。其中Li1.6Mn1.6O4吸附劑的吸附原理為離子交換法:由于Li1.6Mn1.6O4是錳、氧、鋰3種元素形成的晶體,利用H+將晶體中的鋰置換出來,形成具有鋰空位的化合物H1.6Mn1.6O4,該化合物能夠在鋰溶液中將鋰離子重新吸附,而將氫離子重新置換出來。因為吸附原理簡單,吸附劑自身的鋰含量高,因此Li1.6Mn1.6O4和H1.6Mn1.6O4顆粒具有較好的吸附和脫附效果。筆者以KMnO4為原料,經水熱反應、煅燒[8]制備出具有較好吸附效果的尖晶石型鋰錳氧化物Li1.6Mn1.6O4以及H1.6Mn1.6O4,并對其進行了表征,對水中Li+的吸附效果進行了測試。
1 實驗部分
1.1 材料的合成
1.1.1 實驗試劑與設備
試劑:高錳酸鉀、鹽酸、乙醇、氫氧化鋰,均為分析純。設備:磁力攪拌器、水熱反應釜、電熱恒溫鼓風干燥箱、馬弗爐、電子天平、D8-S4型X射線衍射儀、S-4800型掃描電鏡、180-80型原子吸收光譜儀。
1.1.2 γ-MnOOH的制備
將5.0 g KMnO4加入到120 mL蒸餾水中,攪拌,待完全溶解后轉移至200 mL內襯聚四氟乙烯水熱反應釜中。加入10 mL乙醇,混合均勻后繼續添加蒸餾水30 mL。密封,放入140℃烘箱中。24 h后取出反應釜并自然冷卻至室溫。將反應產物過濾,用乙醇和純水洗滌后放入60℃烘箱中干燥4 h。
1.1.3 LiMnO2的制備
在水熱反應釜中配制2mol/LLiOH溶液160mL,加入1.1.2節制備的γ-MnOOH固體8 g,混合均勻。將反應釜放入120℃烘箱中,反應24 h后取出反應釜并自然冷卻至室溫。將制得的產物過濾,用乙醇和純水洗滌后放入60℃烘箱中干燥4 h。
1.1.4 Li1.6Mn1.6O4與H1.6Mn1.6O4的制備
取1.1.3節制得的LiMnO2固體,研磨后放入400℃馬弗爐中燒結4 h。將產物自然冷卻至室溫,得到尖晶石型鋰錳氧化物Li1.6Mn1.6O4固體。將Li1.6Mn1.6O4固體加入到0.5 mol/L HCl溶液中攪拌6 h,使Li+與H+交換,制得具有鋰離子吸附效果的吸附劑H1.6Mn1.6O4。
1.2 吸附與脫附實驗
1.2.1 配制鋰溶液
在吸附與脫附實驗中,為了使合成的吸附劑能夠在較好的條件下吸附,需要配制濃度較高的LiOH溶液。取LiOH固體0.291 g,加入到1 L蒸餾水中,使溶液中Li+質量濃度達到85 mg/L。
為了檢測合成的吸附劑能否對極低濃度的鋰離子進行有效吸附,需要配制與海水鋰離子質量濃度相等的LiOH溶液。取LiOH固體0.011 g,加入到20 L蒸餾水中,使Li+質量濃度達到0.17 mg/L。
1.2.2 吸附過程
將1 g H1.6Mn1.6O4固體顆粒加入到1 L Li+質量濃度為85 mg/L的LiOH溶液中攪拌24 h,并分別在0.5、1.0、2.0、4.0、8.0、16.0、24.0 h 取樣過濾。 對濾液進行原子吸收光譜測試,測定吸附量與時間的關系,并確定所能達到的最大吸附量。
1.2.3 脫附過程
取4份吸附飽和的Li1.6Mn1.6O4固體顆粒各1 g,分別加入到 1 L濃度分別為 0.5、1.0、1.5、2.0 mol/L的鹽酸溶液中攪拌6 h,使固體中的鋰元素與溶液中H+交換,過濾。用原子吸收光譜儀測試濾液中Li、Mn元素的含量,通過測定脫附率和溶損率確定脫附液適宜濃度。
將1 g Li1.6Mn1.6O4固體加入到1 L 0.5 mol/L的鹽酸 溶 液 中攪拌6h,分別在 0.5、1.0、2.0、4.0、6.0 h取樣過濾。對濾液進行原子吸收光譜測試,測定脫附量與時間的關系。
1.2.4 多次吸附-脫附測試
將Li1.6Mn1.6O4固體加入到0.5 mol/L鹽酸溶液中攪拌6 h過濾。將脫附后的固體加入到Li+質量濃度為85 mg/L的LiOH溶液中攪拌12 h過濾。如此反復5次,測試每次濾液中Li和Mn的含量。
1.2.5 低濃度鋰溶液的吸附
取20 L Li+質量濃度為0.17 mg/L的LiOH溶液,加入過量吸附劑H1.6Mn1.6O41 g,攪拌,分別在第7 d與第10 d取樣過濾,測定濾液中Li+質量濃度。
1.3 表征與檢測
用X射線衍射儀對合成的產物進行表征,同時用掃描電鏡對產物形貌進行表征。使用原子吸收光譜儀對溶液中溶解的元素進行測定,確定溶液中目標元素的含量。
2 實驗結果
2.1 材料表征
用X射線衍射儀和掃描電鏡對合成的γ-MnOOH、LiMnO2、Li1.6Mn1.6O4、H1.6Mn1.6O44 種 固 體 進行表征。圖1為4種產物XRD譜圖。從圖1可以看出,各物質衍射峰較為清晰[8],說明每一步反應合成的產物均較純,雜質較少,可以進行下一步合成與測試。圖2為4種產物SEM照片。從圖2可以看出,MnOOH晶體呈棒狀,顆粒較大,平均長度約為7 μm。 LiMnO2、Li1.6Mn1.6O4、H1.6Mn1.6O43 種晶體,從其XRD譜圖和SEM照片可以看出,晶體類型較為相似;并且其晶體較小,約為1 μm,形成的顆粒較細。
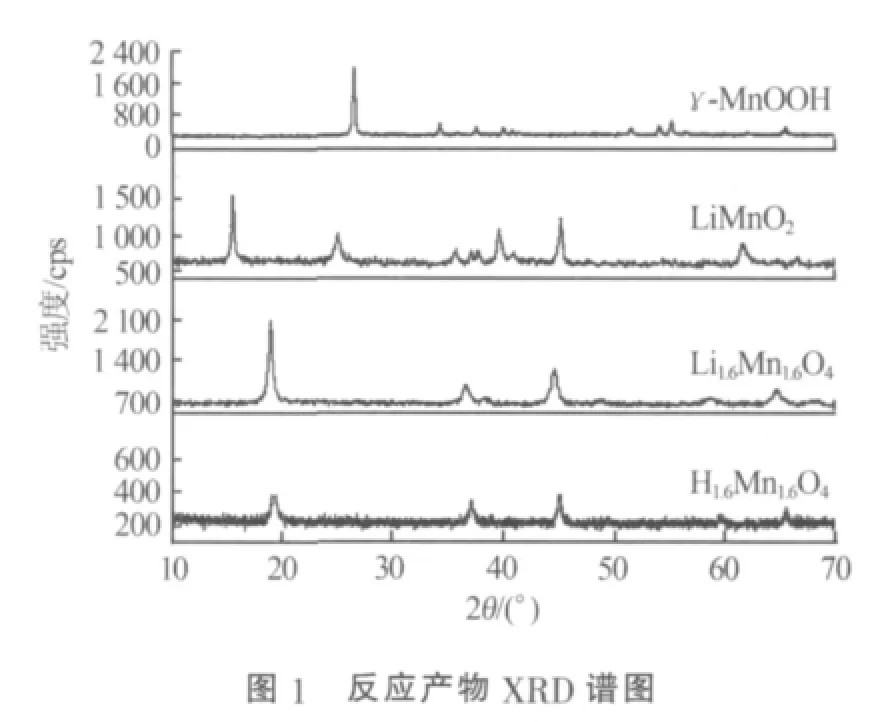

2.2 脫附與吸附過程
2.2.1 吸附時間的測定以及脫附液濃度對脫附效果的影響
將1 g H1.6Mn1.6O4固體加入到1 L Li+質量濃度為85 mg/L的LiOH溶液中,溶液由磚紅色逐漸變成咖啡色。圖3為吸附過程中Li+質量濃度與時間的關系。從圖3可知,溶液中鋰離子質量濃度隨著時間的增加而逐漸降低,大約12 h以后吸附趨于平衡,24 h后Li+質量濃度僅剩39 mg/L。由此可知,合成的吸附劑在Li+質量濃度為85 mg/L情況下能夠達到的最大吸附量為46 mg/g,高于其他文獻報道的尖晶石型吸附劑所能達到的吸附量,如Li1.33Mn1.67O4(吸附量為25.5 mg/g)[11]、LiMn2O4(吸附量為 7.8 mg/g)[12-13]。 這主要是因為合成的Li1.6Mn1.6O4其自身的鋰含量較高,根據尖晶石型吸附劑的吸附原理可知,其對應的理論吸附量也較高。因此,采用該方法合成的吸附劑相對于其他吸附劑在吸附量方面更有優勢。
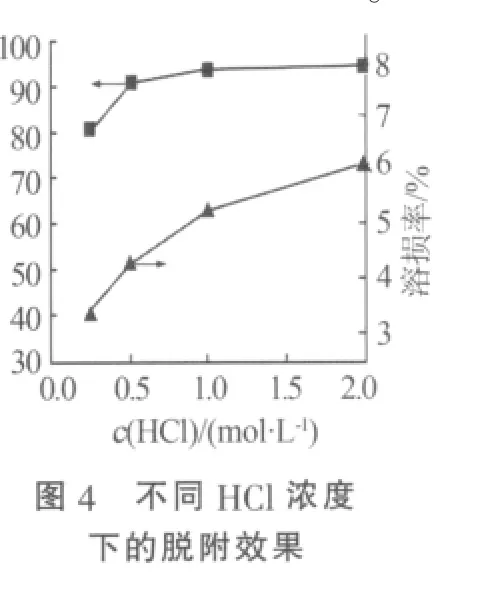
圖4為Li1.6Mn1.6O4在不同濃度的HCl溶液中的脫附率和溶損率的變化。從圖4可知,Li1.6Mn1.6O4的脫附率和溶損率隨HCl濃度的增加而增大。當HCl濃度過低(如H+濃度為0.25 mol/L)時,其溶損率最低,但其脫附效果最差,僅達到82%。當H+濃度大于0.5 mol/L以后,其脫附效果提升不明顯。因此,綜合考慮吸附量和溶損率,選擇鹽酸濃度為0.5 mol/L。
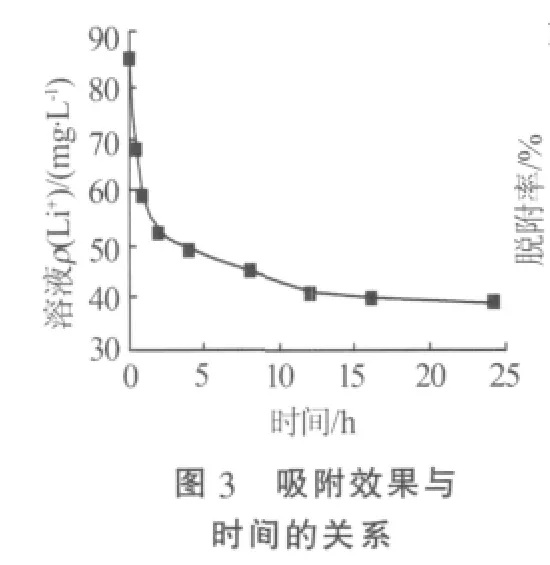
2.2.2 脫附時間的測定以及吸附劑多次吸附-脫附的效果
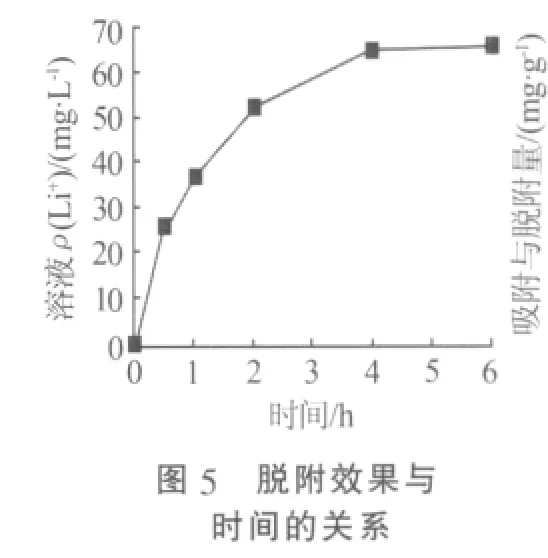
將1 g Li1.6Mn1.6O4固體加入到1 L 0.5 mol/L鹽酸溶液中脫附,溶液由咖啡色逐漸變為磚紅色。圖5是脫附過程中溶液Li+質量濃度與時間的關系。從圖5可知,溶液中Li+質量濃度隨著時間的增加逐漸增大。當脫附時間為6 h時,溶液中Li+質量濃度達到65 mg/L,脫附過程趨于平衡。通過與Li1.6Mn1.6O4固體Li含量對比,可知脫附率已經達到95%以上。
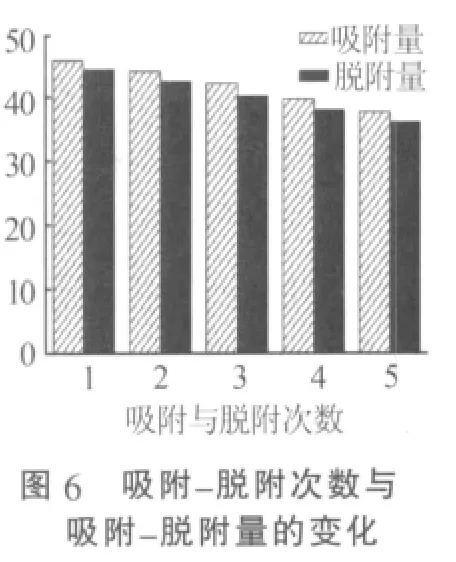
圖6為制備的吸附劑H1.6Mn1.6O4在Li+質量濃度為85 mg/L的LiOH溶液以及0.5 mol/L的HCl溶液中吸附-脫附的次數與脫附-吸附量之間的關系。從圖6可以看出,合成的吸附劑在經過多次吸附-脫附實驗后,其吸附量雖略有下降,但均可以保持高于35 mg/g的吸附量。通過測定溶液中錳元素的含量可知,吸附劑每次脫附的溶損率均在3%以下。說明吸附劑經過多次吸附-脫附實驗均可保持較低的溶損率。由此可知,合成的吸附劑性質比較穩定,可以重復使用,并保持較高的吸附-脫附性能。
2.2.3 低濃度Li+溶液的吸附效果
向20 L低Li+質量濃度(0.17 mg/L)溶液中加入過量的H1.6Mn1.6O4約1 g,檢測其吸附效果。在攪拌條件下,第7 d測得溶液中Li+質量濃度降低為0.04 mg/L,第 10 d 降低為 0.02 mg/L,Li+吸附率可以達到88%。由以上數據可知,合成的吸附劑不僅在高濃度的LiOH溶液中有吸附效果,在極低濃度的Li+溶液中也有吸附效果。
3 結論
以KMnO4為原料,通過水熱法,經氧化還原反應,再經高溫煅燒,然后再在HCl溶液中置換Li+,得到高吸附量的鋰吸附劑H1.6Mn1.6O4。制備的產物經過吸附與脫附實驗表明其有較好的吸附與脫附效果。在吸附過程中,經過12 h可以達到吸附平衡,且最大吸附量可以達到46 mg/g,與其他文獻報道的鋰吸附劑相比吸附量大、吸附平衡時間短。在脫附過程中,在適宜條件下,經過6 h可以達到脫附平衡,脫附率達到95%以上,溶損率僅3%。吸附劑經過反復吸附-脫附實驗,其吸附效果沒有顯著下降。吸附劑在低濃度鋰溶液中也有較好的吸附效果。
[1]冀康平.鋰資源的開發與利用[J].無機鹽工業,2005,37(5):7-9.
[2]袁俊生,紀志永.海水提鋰研究進展[J].海湖鹽與化工,2003,32(5):29-33.
[3]汪鏡亮.鹵水鋰資源提鋰現狀[J].化工礦物與加工,1999,28(12):1-5.
[4]王高尚.鹽湖提鋰技術發展對全球鋰礦業的影響[J].資源產業,2001(5):37-38.
[5]游清治.世界鋰的資源、生產與應用前景[J].世界有色金屬,2008(5):42-45.
[6]袁俊生,孟興智,紀志永.尖晶石型鋰離子篩吸附劑前驅體的合成研究[J].海湖鹽與化工,2005,34(1):6-9.
[7]Dang V D,Steinberg M.Preliminary design and analysis of recovery of lithium from brine with the use of a selective extractant[J].Energy,1978,3(3):325-336.
[8]Ramesh Chitrakar,Hirofumi Kanoh,Yoshitaka Miyai,et al.Recovery of lithium from seawater using manganese oxide adsorbent(H1.6Mn1.6O4) derived from Li1.6Mn1.6O4[J].Ind.Eng.Chem.Res.,2001,40(9):2054-2058.
[9]Kang-Sup Chung,Jae-chun Lee,Wan-keun Kim,et al.Inorganic adsorbent containing polymeric membrane reservoir for the recovery of lithium from seawater[J].Journal of Membrane Science,2008,325(2):503-508.
[10]Wang Lu,Ma Wei,Liu Ru,et al.Correlation between Li+adsorption capacity and the preparation conditions of spinel lithium manganese precursor[J].Solid State Ionics,2006,177 (17/18):1421-1428.
[11]Brett A,Deborah J Jones,Jacques R,et al.Mechanism of proton insertion and characterization of the proton sites in lithium manganate spinels[J].Chem.Mater,1995,7(11):2151-2160.
[12]Aya Umeno,Yoshitaka Miyai,Norio Takagi,et al.Preparationand adsorptive properties of membrane-type adsorbents for lithium recovery from seawater[J].Ind.Eng.Chem.Res.,2002,41(17):4281-4287.
[13]大井健太.海水からのリチヴム采取技術の開發[J].日本海水學會志,1997,51(5):285-288.
猜你喜歡
中學生數理化·八年級物理人教版(2021年12期)2021-12-31 03:23:08
中老年保健(2021年12期)2021-11-30 02:58:01
中學生數理化·中考版(2020年10期)2020-11-27 01:59:48
中國生殖健康(2019年2期)2019-08-23 08:12:08
攝影之友(影像視覺)(2019年2期)2019-03-05 08:27:14
中華詩詞(2018年11期)2018-03-26 06:41:34
產品可靠性報告(2017年7期)2017-09-05 09:49:12
Coco薇(2016年8期)2016-10-09 02:11:50
汽車觀察(2016年3期)2016-02-28 13:16:26
中國醫藥科學(2015年19期)2015-02-27 12:33:11
