油品深度脫硫研究進展
2012-10-26 03:31:32劉旭光趙慧君
太原理工大學學報 2012年3期
劉旭光,趙慧君
(太原理工大學a.化學化工學院;b.新材料界面科學與工程教育部重點實驗室,太原 030024)
隨著工業和交通運輸業的飛速發展,人們越來越關注有害氣體排放造成的空氣污染。燃料中的含硫化合物燃燒變成硫的氧化物排放到大氣中,大量硫的氧化物就是空氣污染的罪魁禍首——溶于雨水則造成腐蝕性極強的酸性降雨,被顆粒物中的鐵、錳催化氧化則形成硫酸霧。治本之策是降低汽油和柴油的硫含量,從源頭上控制污染物排放。美國和歐盟分別限定油品硫含量不得超過15μg/g和10μg/g。我國的北京和上海已先后于2007年和2009年分別制定地方標準,限定汽油、柴油含硫量在50 μg/g以下,提前實施了國Ⅳ標準。針對世界范圍內油品深度脫硫的要求,脫去油品中噻吩類、硫醇、硫醚等含硫物顯得至關重要,但傳統的脫硫方式對于噻吩及其衍生物的脫除又很困難,因此研究新型的環境友好型脫硫技術尤為重要。幾種油品脫硫技術目前實驗室所能達到的指標如表1所示。

表1 實驗室中油品脫噻吩類指標
多年來,對于傳統技術的不斷改進使得深度脫硫有了很大的邁進,但始終無法擺脫其對高溫、高壓、高成本、高能耗、重污染等條件的依賴。表面分子印跡吸附材料作為一種新型油品深度脫硫技術,無毒、穩定、綠色、低成本、低能耗、可再生,具有構效預定性、特異識別性和選擇性,可實現深度脫硫并同時獲得高附加值產品,具有極大的開發潛力和良好的應用前景。
1 傳統脫硫方法
1.1 加氫脫硫
目前通用的燃料油脫硫方法是加氫脫硫(HDS),但該法有明顯的弊端。首先,難以達到深度脫硫的要求,且容易降低燃料油的辛烷值,從而降低汽油的燃燒性能。為了提高辛烷值,需要加入其他化學原料,這樣又提高了成本。此外,加氫脫硫需要高溫高壓加氫,至少是中壓加氫,存在對裝置的要求、安全和加氫成本問題。為了滿足新的硫含量標準,人們加大了工業化加氫脫硫和非加氫脫硫的研究力度。Schmitz等[9]采用一種兩相反應器實現了加氫深度脫硫,預飽和處理器中的H2含量遠超過已加氫油品加氫脫硫所需用量,不再需要用氣態H2向液相進行循環補充,從而省去了傳統加氫脫硫滴流床反應器所必需的H2供給循環裝置,取而代之的是相對簡單的液體循環裝置,降低了設備成本。針對加氫深度脫硫的油品辛烷值和油品收率問題,各大公司以及研究機構爭相開發多效催化劑[10],如介孔分子篩材料以其獨特的孔道結構和孔徑分布、高的比表面積及較好的熱穩定性和水熱穩定性等特點,近年來被廣泛地用作油品深度加氫脫硫催化劑載體[11]。但改進的加氫脫硫技術仍然要求較高的溫度和壓力、活性更高的催化劑和更長的反應時間,相應地增加了操作成本,學者們也更多地轉向了對非加氫脫硫技術的研究。
1.2 氧化脫硫
氧化脫硫技術(ODS)操作條件溫和,選擇性高,且不需要氫氣,降低了操作成本,成為近十幾年來研究較多、發展最快的非加氫脫硫技術。氧化脫硫的原理是:用氧原子把油品中的含硫化合物氧化,在氧化劑作用下,不斷促使硫化物氧化、降解,從而達到脫硫的目的。H2O2是一種強氧化劑,脫硫唯一的副產品是水,易于除去,是使用最普遍的氧化劑[12]。Asghar等[13]用H2O2作氧化劑,甲酸為催化劑,在溫和的反應條件下脫硫30min,隨后進行液液萃取,除硫率可達到87%。Duarte等[1]利用一定比例的H2O2∶HAC加以超聲處理,以甲醇作為萃取液,對于模擬油品(溶質為二苯并噻吩(DBT)和4,6-二甲基苯并噻吩(4,6-DMDBT))和柴油的脫硫率分別為98%和75%,與常規的氧化脫硫相比,可以用更少量的H2O2、醋酸和甲醇,減少了反應時間。Jiang等[14]在H2O2作氧化劑且在溫和的反應條件下加入一種乳狀物催化劑,在乳狀物介質中由于相間傳質而產生局限,兩親乳狀物催化劑能有選擇地氧化柴油中的含硫分子生成相應的砜類,可通過極性萃取對其進行移除,含硫量可從幾百μg/g降至0.1μg/g(處理加氫預處理過的柴油),若改用直餾柴油,其含硫量可從6000μg/g降到30μg/g。張存等[15]在 H2O2/WO3/ZrO2氧化體系中對以甲苯為溶劑、DBT為典型含硫化合物的模擬油品進行了氧化脫硫研究,考察了反應溫度、反應時間、氧化劑加入量、催化劑用量對DBT轉化率的影響,在WO3/ZrO2固體超強酸催化作用下,H2O2氧化DBT較易進行,此時DBT轉化率達到96%以上。但氧化脫硫是將硫化物轉化成具有毒性的砜類,廢液處理不當會造成嚴重的環境污染。
1.3 催化脫硫
在氧化脫硫技術的基礎上逐步添加一定的金屬及金屬化合物類催化劑,結合催化氧化作用,對油品進行催化脫硫(CDS)。Yuan等[2]在具有氧化作用的超臨界水反應器中利用催化劑CoMo/γ-Al2O3進行催化氧化脫硫研究,對以苯并噻吩(BT)為溶質的模擬油品脫硫量可達到67%。Prasad等[16]通過擔載MoO3的催化劑進行脫硫實驗,300h之后仍表現出穩定的催化活性。Liu等[17]利用環境友好型Fe(Ⅵ)系催化氧化劑對柴油進行深度脫硫實驗,Fe(Ⅵ)的還原與Cr、Mn不同,會轉化成一種相對無毒的Fe(Ⅲ)化物,催化氧化后用糠醛萃取,其硫的移除率可達到96.7%。Yan等[18]利用Ag修飾的介孔HPW/SiO2催化劑對直餾柴油催化氧化脫硫,脫硫量達到87.3%。隨后出現了光催化氧化脫硫技術[19],光催化氧化脫硫可以在室溫常壓下進行,將燃料油中的含硫化合物氧化成極性物質后,很容易結合萃取、吸附、蒸餾等方法除去氧化產物,最終可實現油品中硫含量為20μg/g的目標。但要想實現紫外光的工業化應用,仍具有極大的局限性,并且催化法脫硫效率雖較高,但在催化劑上的投資較大,制備條件又苛刻,出于經濟效益的考慮,仍需要很長的發展歷程。
1.4 萃取脫硫
順著油品清潔化趨勢的發展浪潮,又提出了離子液體萃取法[20],離子液體(ILs)是在室溫下處于液態的熔鹽,與有機液體相比,離子液體具有不揮發、不腐蝕的優點,且對噻吩類物質具有較好的萃取能力,適合于萃取脫硫(EDS)。利用離子液體把汽油中的硫化物萃取出來,并加以利用,剩余的很少一部分硫化物可以用氧化法去除。寧英男等[21]利用AlCl3、FeCl3和CuCl與氯代丁基甲基咪唑反應合成三種離子液體,分別用于汽油和柴油的脫硫試驗,結果表明AlCl3型離子液體的脫硫效果最好,對汽油和柴油的脫硫率分別為89.5%和58.24%,其中AlCl3型和FeCl3型兩種離子液體在常溫下均具有良好的穩定性和流動性,易與脫硫后的汽油或柴油分離,具有良好的應用前景。應用于油品深度脫硫研究的離子液體種類還有親水性離子液體[22]和酸性離子液體[3]等。近年來,也出現了離子液體萃取法和光催化氧化法相結合的離子液體萃取-光催化氧化燃料油脫硫方法,利用氧化劑和光催化劑將燃料油中的含硫化合物氧化為極性物質,轉移到離子液體中,從燃料油中除去,最終可以使汽油中的硫含量降到10μg/g。但是離子液體的制備成本一般比較高,因此在合成過程中仍需進一步降低原料成本,簡化合成過程,增強溶劑和原料的循環利用,降低環境污染,提高反應轉化率等,發展遠景很好。
1.5 吸附脫硫
吸附法脫硫(ADS)的基本原理是利用固體吸附劑選擇性吸附含硫有機化合物,從而實現將硫化物從油品中脫除的目的。吸附脫硫的關鍵在于吸附劑的選擇性、吸附容量、再生能力以及達到良好吸附效果的操作條件。根據吸附劑與含硫化合物作用機理的不同,ADS可分為反應吸附脫硫(RADS)和選擇性吸附脫硫(SADS)。RADS是指吸附劑的活性金屬組分與含硫化合物的硫原子發生化學反應,硫以金屬硫化物的形式留在吸附劑上,剩下的烴類部分返回油品中。Huang等[23-24]研究了 Ni/ZnO作為吸附劑在反應吸附脫硫應用中的反應過程及在不同氣氛下的脫硫機理,柴油中的有機硫化物在Ni/ZnO催化劑的Ni表面分解生成Ni3S2,并在H2存在條件下生成H2S,進而通過與ZnO作用轉化成ZnS,最終儲存在吸附劑中;而在N2氣氛中,是物理和化學選擇性吸附,脫硫能力相對較低。SADS是基于固體吸附劑對有機硫化合物的選擇吸附能力而開發的一類燃料油脫硫技術[25],一般在低溫、常壓下進行,操作條件溫和,再生可通過脫附溶劑清洗或還原性氣體吹掃來實現。如何從含有大量不飽和烴類競爭吸附分子的復雜油品體系中選擇性地脫除含硫化合物是開發SADS工藝的難點。
1.6 烷基化脫硫
烷基化脫硫技術主要用于脫除流化催化裂化(FCC)汽油中的噻吩類化合物。FCC汽油中的噻吩硫化合物在酸性催化劑的作用下與烯烴進行烷基化反應,生成沸點較高的烷基噻吩化合物,然后利用沸點的差別進行分餾脫除,這樣即可脫除汽油中的硫化物,又可降低烯烴含量。該法以磷酸、硫酸、硼酸、氫氟酸、BF3、BCl3、FeCl2等為酸性催化劑,以氧化鋁、氧化硅、硅藻土等為載體。酸性對噻吩轉化率的影響很明顯。酸性催化劑的孔分布對噻吩與烯烴進行烷基化反應生成的高沸點化合物的沸點也有影響,介孔分子篩有利于形成高沸點的烷基化產物。張繁軍等[26]以溴乙烷為烷基化劑、四氟化硼鉀為沉淀劑,進行烷基化沉淀脫硫,對模擬含硫輕質油品總硫脫除率達到76.5%。DBT類化合物中的硫在常溫下被烷基化生成易結晶的锍鹽。锍鹽的極性非常高,幾乎不溶于無極性的碳氫溶劑。此反應用于輕質油品的脫硫,可使油品中其他方法很難脫除掉的DBT類以锍鹽形式沉淀而分離。但該技術需依賴強酸性催化環境,極大地提高了反應設備的多方面指標。
1.7 輻射誘導脫硫
采用γ射線對油品進行輻射處理,含氧空氣被電子相互轟擊產生的能量激發而充當氧化劑,將油品中的硫醇等含硫化合物中的硫重新分配,轉化成為砜、SOx、酸等,可用于油品脫硫的進一步深入研究[27]。Basfar等[28]利用γ射線使樣品的微觀結構發生改變,有利于進而通過常規方法進行油品脫硫。對于原油和直餾柴油,將γ射線與其他的物理/化學過程相結合而實現溫和條件深度油品脫硫,具有巨大的潛在應用價值。
1.8 生物脫硫
生物脫硫(BDS)是指利用一系列酶催化反應在溫和的條件下脫除化石燃料中的含硫化合物的過程。生物脫硫具有選擇性高、副反應少、反應條件溫和、投資少、對燃料熱值影響小等優點。雖然該法技術還不成熟,許多機理有待進一步研究,但已成為令人矚目的綠色脫硫技術,引起了眾多科學家和工程師的興趣,預計有可能成為未來脫硫技術的研究方向之一[4]。對于脫硫所用微生物的培養、壽命、后續處理等問題仍需深入研究。
2 表面分子印跡脫硫
分子印跡技術指制備對某一特定的目標分子(也稱模板分子,印跡分子)具有特異預定選擇性的聚合物的一項技術。所制備的聚合物稱為分子印跡聚合物(MIPs)。由于具有構效預定性、特異識別性和廣泛實用性等優點,被廣泛應用于色譜分離、固相提取、生物傳感器、選擇性催化等眾多領域的研究中[5,29-33]。
MIPs對印跡分子的特異識別功能正是源于它擁有大量與印跡分子空間立體結構相吻合、作用位點一一對應的印跡孔穴。當MIPs在適當的介質中遇到模板分子時就會發生特異性識別作用。MIPs的傳統制備方法[34-38]所得的印跡聚合物均是無定形的顆粒或微米級的微球,但其識別位點大都包埋在聚合物微球內部,這就對后續的傳質過程造成了阻礙,模板需要克服內部阻力進行識別,導致待印跡分子與識別位點結合困難,結合速率低,洗脫效率不高,降低MIPs的結合容量和選擇性,因此把識別位點建立在基質表面的表面印跡技術日益受到重視。
表面分子印跡技術,作為一種新型的MIPs的制備方法,與本體聚合、懸浮聚合以及原位聚合相比具有不可比擬的優勢[38]。在固體表面進行烙印,制備過程中模板分子更易洗脫,識別過程中模板分子無需克服內部傳質阻力,這就提高了MIPs的選擇性和吸附速率。目前以K2Ti4O9、TiO2納米球、碳微球(CMSs)等為基質制備了吸附DBT的表面印跡吸附材料,為脫除含硫化合物提供了一條新的途徑,不但可以達到深度脫硫的目的,并且DBT的結構不會被破壞,可實現其綠色應用,這為進一步研究油品的深度脫硫以生產清潔燃料提供了一種新的方法,有廣闊的發展前景。
2.1 K2Ti4O9基表面分子印跡脫硫
Yang等[6]選擇常作催化劑使用的K2Ti4O9充當支撐基質,應用表面分子印跡技術并選用合適的溶劑將其從所制得的K2Ti4O9基表面分子印跡聚合物中去除,最終獲得一種對DBT具有一定選擇識別性的中空型表面分子印跡脫硫材料:首先,用HCl將K2Ti4O9活化,接著用一定量的硅烷偶聯劑3-(甲基丙烯酰氧)丙基三甲氧基硅烷(MPS)修飾活化后的K2Ti4O9,制得 MPS-K2Ti4O9,同時將功能單體乙烯基吡啶(4VP)和模板分子DBT溶于甲苯中,通N2進行凈化處理,反應一段時間后加入MPS-K2Ti4O9、引發劑偶氮二異丁腈(AIBN)和交聯劑乙二醇二甲基丙烯酸酯(EGDMA),獲得K2Ti4O9基表面分子印跡聚合物(S-MIP);然后,在反應器中加入一定量的S-MIP,加入適量丙酮提高疏水顆粒的潤濕性,在HF水溶液中冰浴、攪拌反應以除去基質K2Ti4O9,經過洗滌、干燥等步驟,最終獲得中空型表面分子印跡聚合物H-MIP,利用相同過程不加模板DBT制備中空型非印跡聚合物(HNIP)作為吸附脫硫實驗的對照樣品。吸附性能評價實驗所得數據表明:H-MIP的比表面積為245.15m2/g,平均孔徑3.44nm,對 DBT 含量為500mg/L的模擬油品吸附平衡時間是3h,最大吸附量19.31mg/g,即對DBT的脫除率為9.7%并具有良好的選擇性吸附性能。該研究由于想使對吸附過程的影響最小化,而提出了將表面分子印跡聚合物的基質移除,以制備中空型表面分子印跡脫硫劑的方法,但同時在一定程度上影響了吸附容量。
2.2 TiO2納米球基表面分子印跡脫硫
納米TiO2作為多功能材料引起了越來越多的關注。這種新的無機材料具有許多優秀性質,例如無毒、易得、耐光、低成本、化學穩定,并具有高光催化效率。隨著用表面分子印跡技術結合移除基質過程來制備中空型功能材料理念的逐漸加強,Xu等[7]選用TiO2納米球作為基質,用與制備K2Ti4O9基中空型表面分子印跡聚合物相同的化學試劑和相似的制備過程,最終獲得中空型表面分子印跡聚合物,比表面積為365.80m2/g,平均孔徑為1.69nm。吸附性能表明:模擬油品硫含量由398.89mg/L,4h后降低到323.77mg/L,經過多次吸附過程可降低到250.99mg/L,即對DBT的脫除率為22.5%,吸附行為遵循擬二級動力學模型,用Freundlich等溫線模型等夠比較好地描述吸附平衡數據,熱力學參數顯示其是自發吸熱吸附,對正辛烷中的DBT具有良好的選擇性吸附性能。
2.3 CMSs基表面分子印跡脫硫
CMSs化學穩定性好,熱穩定性高,耐酸堿,導電、導熱性能優良,又由于其制備原料來源廣泛、附加值高,已被用作高密高強碳材料[39]、催化劑載體[40]、超高比表面積活性炭[41]和鋰離子二次電池負極材料[42]等,成為一種具有極大開發潛力和應用前景的碳材料。該類材料自身的結構特征決定了其在功能化應用過程中的優勢:表面修飾后具有豐富的鍵合位,便于分子接枝或錨定;具有很好的酸堿穩定性、熱穩定性和力學性能穩定性。在這些優勢的基礎上,碳球經表面分子印跡獲得對較大尺寸的分子的識別與選擇性吸附功能,無疑具有極為廣闊的用途,故CMSs表面分子印跡材料的研究具有很好的理論價值和應用前景。
筆者課題組所研究的表面分子印跡脫硫技術正是以物理、化學性質優良的CMSs為基質,DBT為模板,通過酸化、硅烷化、接枝聚合、溶劑洗脫等過程制備對DBT分子具有特異性、選擇性識別吸附性能的CMSs基表面分子印跡吸附材料(如圖1),目前主要通過三種不同途徑獲得了實質性的脫硫效果。

圖1 表面分子印跡材料的制備工藝
2.3.1 原位聚合法合成表面分子印跡材料(MIPPMAA/CMSs)
以DBT為模板分子、氯仿為溶劑、甲基丙烯酸(MAA)為功能單體,采用原位聚合法制備CMSs表面DBT分子印跡聚合物(MIP-PMAA/CMSs)[8]。
首先,取0.5g混酸(濃 HNO3/H2SO4,體積比1∶3)氧化CMSs加入200mL乙醇與水的混合溶劑(體積比為1∶1)中,并加入10mL kH-570,通氮氣,在55℃反應14h,抽濾、洗滌、分離、干燥,制得硅烷化CMSs。然后,將0.369g DBT溶于20mL氯仿中,置于三口燒瓶,再加入0.3g硅烷化CMSs和1mL MAA,攪拌30min后加入0.065g引發劑AIBN和4mL交聯劑乙二醇丙烯酸乙二醇酯(EDMA),將水浴溫度調節至60℃加熱24h。聚合完成后,用乙醇洗滌以除去物理吸附在CMSs表面的聚合物。再用體積比為9∶1的甲醇和乙酸混合溶液洗滌直到除去模板DBT,最后干燥,制得 MIPPMAA/CMSs。另外,制備不含模板分子的非印跡聚合物(NIP-PMAA/CMSs)。
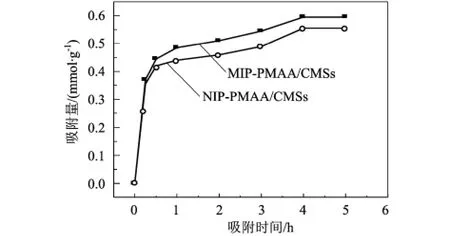
圖2 MIP-PMAA/CMSs和NIP-PMAA/CMSs的吸附動力學曲線(室溫,DBT:8mmol/L)
從吸附DBT分子的吸附動力學曲線(圖2)可以看出,隨著吸附時間的延長,MIP-PMAA/CMSs和NIP-PMAA/CMSs對DBT的吸附量增加;當吸附時間增至5h時,吸附量基本趨于平衡。
0.1 g的 MIP-PMAA/CMSs或 NIP-PMAA/CMSs加入到20mL不同濃度的DBT的正己烷溶液中,振蕩時間為6h,測得吸附量,繪制的等溫吸附曲線如圖3所示。可以看出,濃度增大時,MIPPMAA/CMSs對模板DBT的吸附量隨之增大,當濃度增加到一定程度時,吸附量達到飽和。而NIPPMAA/CMSs的吸附量較 MIP-PMAA/CMSs更快達到飽和,對于相同濃度的DBT,MIP-PMAA/CMSs的吸附量明顯高于NIP-PMAA/CMSs的吸附量。
2.3.2 引發轉移終止接枝聚合法合成(MIPPMAA/CMSs)
引發轉移終止劑(Iniferter)聚合是一種活性自由基聚合方法,在自由基聚合過程中同時起到引發、轉移和終止作用[43]。Iniferter聚合可避免凝膠化反應,聚合條件溫和,工藝簡單。筆者課題組采用引發轉移終止劑聚合法合成DBT印跡的MIP-PMAA/CMSs。

圖3 MIP-PMAA/CMSs和NIP-PMAA/CMSs的等溫吸附曲線(室溫,吸附6h)
首先,利用Iniferter修飾CMSs:將0.4g混酸氧化CMSs置于三口燒瓶中,加入25mL甲苯,再量取0.45mL p-(氯甲基)苯基三甲氧基硅烷(CMTMS)溶于10mL甲苯中,加入三口燒瓶,60℃攪拌4h后冷卻、抽濾、洗滌,收集抽濾產品,烘干后得到硅烷化CMSs。然后,稱0.3g硅烷化CMSs和20mL甲苯加至三口燒瓶,再加入溶有0.046g二乙基二硫代氨基甲酸鈉(DDTC)的乙醇溶液10 mL,超聲分散,于25℃水浴中加熱12h;抽濾、洗滌、烘干得到Iniferter修飾的CMSs。
其次,CMSs表面接枝PMAA:0.2g經Iniferter修飾的CMSs和20mL乙醇放入三口燒瓶,加MAA,在紫外燈光照下反應,置涼后抽濾、洗滌、烘干得到PMAA/CMSs復合物。
最 后,合 成 MIP-PMAA/CMSs:將 0.1g PMAA/CMSs分散于20mL的氯仿中,加至三口燒瓶,加入0.1843g DBT;超聲分散后,25℃條件下攪拌3h后加入4mL EDMA,60℃水浴加熱24 h;冷卻后抽濾,并依次用氯仿和乙醇反復洗滌,收集產品,烘干;再用無水甲醇和冰乙酸(體積比9∶1)的混合溶液洗脫,烘干得到 MIP-PMAA/CMSs。另外,也制備了非印跡聚合物(NIP-PMAA/CMSs)。
從吸附動力學曲線(圖4)看出,MIP-PMAA/CMSs對DBT的吸附量明顯高于NIP-PMAA/CMSs,說明其對DBT具有特異識別性。
從MIP-PMAA/CMSs對聯苯(DIP)的動力學吸附曲線(圖5)可以看出,吸附時間達到3h后,MIP-PMAA/CMSs對聯苯的最大吸附量與其對DBT的吸附量相差較大,說明其對模板分子有專一選擇性。
2.3.3 接枝聚合法合成表面分子印跡材料(MIPPAMPS/CMSs)

圖4 MIP-PMAA/CMSs和NIP-PMAA/CMSs的吸附動力學曲線(室溫,DBT:0.8mmol/L)

圖5 MIP-PMAA/CMSs的吸附動力學曲線(室溫,DBT:0.8mmol/L,DIP:0.8mmol/L)
AMPS分子中的-SO3H具有很強的極性,易與模板分子DBT自組裝,形成穩定的單體-模板復合物;且很好地溶解于水中,易實現水相聚合;干燥的AMPS化學穩定性高,不會發生自聚。
以DBT為模板分子,以AMPS為功能單體,同樣經CMSs表面活化、硅烷化、接枝單體、交聯聚合等過程,采用接枝聚合法在CMSs表面合成DBT分子印跡材料(MIP-PAMPS/CMSs)[44,45]。
首先利用KH-570對混酸氧化CMSs進行改性得到硅烷化CMSs。然后,在反應溫度70℃、反應時間12h、AMPS用量1.0g/0.2g硅烷化 CMSs、引發劑質量分數為3%,在硅烷化CMSs表面優化接枝PAMPS。最后,合成 MIP-PAMPS/CMSs:將0.111g的DBT溶于10mL的氯仿,置于三口燒瓶;將三口燒瓶放入集熱式恒溫加熱磁力攪拌器中,加入0.1g PAMPS/CMSs,攪拌30min;加入3mL的交聯劑EDMA,在50℃水浴回流10h;交聯后,用甲醇和乙酸(體積比9∶1)的混合溶液離心以洗脫聚合物表面的DBT;傾掉上清液,將剩余物質50℃下烘干,得到 MIP-PAMPS/CMSs。同樣也制備了無DBT印跡的NIP-PAMPS/CMSs。動態吸附(表2,3)發現:0.1g MIP-PAMPS/CMSs在通過3mL、1mmol/L DBT溶液后達到飽和,吸附量為1.38×10-2mmol/g;0.1g NIP-PAMPS/CMSs在通過1 mL、1mmol/L DBT溶液后就達到飽和,吸附量僅為 1.66×10-3mmol/g。MIP-PAMPS/CMSs 對DBT的選擇性識別遠優于NIP-PAMPS/CMSs。

表2 MIP-PAMPS/CMSs動態吸附量

表3 NIP-PAMPS/CMSs動態吸附量
3 結束語
在綜述脫硫技術發展的基礎上,研究一種新型CMSs表面分子印跡脫硫技術,憑借其無毒、穩定、綠色、低成本、低能耗、可再生、高選擇性,可實現油品深度脫硫,并同時獲得高附加值產品,展現出其巨大的發展空間及廣闊的應用領域,預計會成為油品深度脫硫技術重要的研究方向之一。
1)利用原位聚合法合成表面分子印跡材料,MIP-PMAA/CMSs可選擇性識別DBT,對其最大吸附量為0.595mmol/g,脫硫率達73.5%;
2)利用引發轉移終止接枝聚合法合成表面分子印跡材料,MIP-PMAA/CMSs和 NIP-PMAA/CMSs對DBT的飽和吸附時間為3h,最大吸附量分別為0.4821mmol/g和0.2416mmol/g,表明其對DBT具有良好的選擇性吸附能力。
3)利用接枝聚合法合成表面分子印跡材料,MIP-PAMPS/CMSs吸附 DBT飽和量為1.38×10-2mmol/g,對于 DBT吸附能力要高于 NIPPAMPS/CMSs(1.66×10-3mmol/g)。相比之下,該法合成的表面分子印跡材料對DBT的吸附容量較小,可能是由于選用的功能單體在接枝聚合法中與模板分子的作用能力不強,所得到的印跡CMSs對DBT的印跡空穴不豐富,對此,本課題組正在嘗試對功能單體的優選研究。
4)目前,為制備高吸附性能的表面分子印跡材料,本課題組正在嘗試利用可逆加成-斷裂鏈轉移法和多孔碳為基質來合成:可逆加成-斷裂鏈轉移法可在聚合體系中活性鏈自由基控制在低濃度時不影響鏈增長反應,而雙分子偶合或歧化終止等副反應減少,控制聚合,使表面分子印跡過程更有可控性;多孔球形碳材料與光滑CMSs相比,由于孔的出現,比表面積增大而產生表面效應,反應活性增強,可望在印跡過程中提高基質的溶劑相容性和表面活性以達到增強印跡效果的目的。
[1]Fábio A Duarte,Paola de A Mello,Cezar A Bizzi,et al.Sulfur removal from hydrotreated petroleum fractions using ultrasound-assisted oxidative desulfurization process[J].Fuel,2011,90(6):2158-2164.
[2]Yuan P Q,Cheng Z M,Jiang W L,et al.Catalytic desulfurization of residual oil through partial oxidation in supercritical water[J].J Supercritical Fluids,2005,35(1):70-75.
[3]張存,王峰,潘小玉,等.酸性離子液體萃取-氧化模擬油品脫硫研究 [J].燃料化學學報,2011,39(9):689-693.
[4]羅明芳,高紅帥,李玉光,等.油品固定化細胞脫硫研究進展 [J].化工進展,2009,28(11):1986-1990.
[5]Liu X J,Ouyang C B,Zhao R,et al.Monolithic molecularly imprinted polymer for sulfamethoxazole and molecular and molecular recognition properties in aqueous mobile phase[J].Analytica Chimica Acta,2006,571(2):235-241.
[6]Yang W M,Liu LK,Zhou W,et al.Preparation and evaluation of hollow molecular imprinted polymer for adsorption of dibenzothiophene[J].Appl Surf Sci,2012.
[7]Xu W Z,Zhou W,Xu P P,et al.A molecularly imprinted polymer based on TiO2as a sacrificial support for selective recognition of dibenzothiophene[J].Chem Eng J,2011,172(1):191-198.
[8]Yang Y Z,Liu X G,Guo M C,et al.Molecularly imprinted polymer on carbon microsphere surface for adsorbing dibenzothiophene[J].Colloids Surf A,2011,377(1-3):379-385.
[9]Schmitz C,Datsevitch L,Jess A.Deepdesulfurization of diesel oil:kinetic studies and process-improvement by the use of a two-phase reactor with pre-saturator[J].Chem Eng Sci,2004,59(14):2821-2829.
[10]葛暉,李學寬,秦張峰,等.油品深度加氫脫硫催化研究進展 [J].化工進展,2008,27(10):1490-1497.
[11]任艷群,王沖,莫家樂,等.介孔分子篩載體在油品深度加氫脫硫中的應用研究進展 [J].化工進展,2011,30(4):743-752.
[12]路文娟,楊延釗.過氧化氫用于油品氧化脫硫的研究進展 [J].化工進展,2009,28(4):605-609.
[13]Dehkordi A M,Kiaei Z,Sobati M A.Oxidative desulfurization of simulated light fuel oil and untreated kerosene[J].Fuel Process Technol,2009,90(3):435-445.
[14]Jiang Z X,Lv H Y,Zhang Y N,et al.Oxidative desulfurization of fuel oils[J].Chin J Catal,2011,32(5):707-715.
[15]張存,王洪娟,劉濤,等.模擬油品氧化脫硫及反應動力學研究 [J].燃料化學學報,2011,39(8):611-614.
[16]Prasad V.V.D.N.,Jeong K E,Chae H J,et al.Oxidative desulfurization of 4,6-dimethyl dibenzothiophene and light cycle oil over supported molybdenum oxide catalysts[J].Catalysis Communications,2008,9(10):1966-1969.
[17]Liu S Z,Wang B H,Cui B C,et al.Deep desulfurization of diesel oil oxidized by Fe(VI)systems[J].Fuel,2008,87(3):422-428.
[18]Yan X M,Shen S G,Lin X.Oxidative desulfurization of diesel oil over Ag-modified mesoporous HPW/SiO2catalyst[J].J Fuel Chem Technol,2009,37(3):318-323.
[19]馬四國,劉翠微,周二鵬,等.輕質油品光催化氧化脫硫研究進展 [J].河北化工,2005(4):4-6.
[20]劉章勇,張玉貞,張小英,等.輕質油品溶劑萃取脫硫技術研究 [J].應用化工,2009,38(7):1067-1072.
[21]寧英男,趙秀麗,張長寶,等.離子液體在輕質油品脫硫中的應用 [J].煉油技術與工程,2008,38(3):27-31.
[22]于穎敏.親水性離子液體在油品脫硫中的應用 [J].曲阜師范大學學報,2009,35(3):64-68.
[23]Huang L C,Wang G F,Qin Z F,et al.A sulfur K-edge XANES study on the transfer of sulfur species in the reactive adsorption desulfurization of diesel oil over Ni/ZnO[J].Chem Commun,2010,11(7):592-596.
[24]Huang L C,Wang G F,Qin Z F,et al.In situ XAS study on the mechanism of reactive adsorption desulfurization of oil product over Ni/ZnO [J].Appl Catal,B:Environ,2011,106(1-2):26-38.
[25]張景成,柳云騏,安高軍,等.吸附脫硫技術生產清潔油品 [J].化學進展,2008,20(11):1834-1845.
[26]張繁軍,韓冬云,曹祖賓,等.模擬輕質油品烷基化沉淀法脫硫 [J].石油學報,2009,25(4):596-599.
[27]Zaykina R F,Zaykin Yu A,Mamonova T B,et al.Radiation methods for demercaptanization and desulfurization of oil products[J].Radiat Phys Chem,2002,63(3-6):621-624.
[28]Basfar A A,Mohamed K A.Radiation-induced desulfurization of Arabian crude oil and straight-run diesel[J].Radiat Phys Chem,2011,80(11):1289-1290.
[29]Chen Y,Kele M,Sajonz P,et al.Influence of thermal annealing on the thermodynamic and mass-transfer kinetic properties of D-and L-phenylalanine anilide on imprinted polymeric stationary phases[J].Anal Chem,1999,71(5):928-938.
[30]Kempe M,osbach K.Separation of amino acids,peptides and proteins on molecularly imprinted stationary phases.J Chro-matogr A,1995,691(1-2):317-323.
[31]Bereli N,Andac M,Baydemir G,et al.Protein recognition via ion-coordinated molecularly imprinted supermacroporous cryogels[J].J Chromatogr A,2008,1190(1-2):18-26.
[32]Mayes A G,Mosbach K.Molecularly imprinted polymer beads:suspension polymerization using a liquid perfluorocarbon as the dispersing phase[J].Anal Chem,1996,68(21):3769-3774.
[33]王妍,荊濤,包學偉,等.本體聚合法制備2-氯酚分子印跡聚合物及其性能評價 [J].分析科學學報,2008,24(5):531-534.
[34]Ansell R J,Mosbach K.Molecularly imprinted polymers by suspension polymerisation in perfluorocarbon liquids,with emphasis on the influence of the porogenic solvent[J].J Chromatogr A,1997,787(1-2):55-66.
[35]Matsui J,Fujiwara K,Ugata S,et al.Solid-phase extraction with a dibutylmelamine-imprinted polymer as triazine herbicide-selective sorbent[J].J Chromatogr A,2000,889(1-2):25-31.
[36]尹俊發,楊更亮,張軼華,等.原位聚合那格列奈分子印跡手性固定相的分子識別特性研究[J].化學學報,2004,62(19):1922-1926.
[37]Gao B J,Wang R X.A comparative study on effects of two kinds of polymerization methonds on grafting of polymer onto silica surface[J].J Appl Polym Scienc,2006,102(6):5808-5817.
[38]李莎.碳微球表面二苯并噻吩分子印跡聚合材料制備過程優化及吸附性能初探[D].太原:太原理工大學,2011.
[39]Wang Y G,Korai Y,Mochida I.Carbon disc of high density and strength prepared from synthetic pitch-derived mesocarbon microbeads[J].Carbon,1999,37(7):1049-1057.
[40]Liu Y C,Qiu X P,Huang Y Q,et al.Methanol electro-oxidation on mesocarbon microbead supported Pt catalysts[J].Carbon,2002,40(13):2375-2380.
[41]呂永根,凌立成,劉朗,等.中間相炭微球的活化 [J].煤炭轉化,1999,22(2):66-70.
[42]Alcantara R,Fernandez M J,Lavela P,et al.Characterisation of mesocarbon microbeads(MCMB)as active electrode material in lithium and sodium cells[J].Carbon,2000,38(7):1031-1041.
[43]Lee H Y,Kim B S,et al.Grafting of molecularly imprinted polymers on iniferter-modified carbon nanotube[J].Biosens Bioelectron,2009,25(3):587-591.
[44]Yang Y Z,Zhang Y,Li S,et al.Grafting molecularly imprinted poly(2-acrylamido-2-methylpropanesulfonic acid)onto the surface of carbon microspheres[J].Applied Surface Science,2012.doi:10.1016/j.apsusc.2012.03.058
[45]李莎,段菲菲,楊永珍,等.硅烷偶聯劑修飾碳微球 [J].功能材料,2011,1(42):25-29.
