氯化鉀與磷酸復分解反應的動力學研究*
2012-11-10 01:02:10付全軍王辛龍傅玉信張志業
無機鹽工業 2012年4期
關鍵詞:實驗
茍 蘋,付全軍,王辛龍,楊 林,傅玉信,張志業
(四川大學化學工程學院,四川成都 610065)
氯化鉀與磷酸復分解反應的動力學研究*
茍 蘋,付全軍,王辛龍,楊 林,傅玉信,張志業
(四川大學化學工程學院,四川成都 610065)
研究了以氯化鉀和熱法磷酸為原料,采用復分解法制備磷酸二氫鉀的方法。考察了反應時間、反應溫度、原料配比對氯化鉀轉化率的影響。在反應溫度為373~413K和反應時間為1~4 h的條件下,研究氯化鉀與熱法磷酸的宏觀反應過程,自阻化作用對該反應過程的反應速率影響較大,阻緩系數β隨反應溫度升高而增大。應用德羅茲多夫方程擬合實驗數據,確定了動力學方程:1/t ln[1/(1-X)]-βX/t=exp(-65 840/RT)+2.215,并求得了反應的表觀活化能Ea=65.84 kJ/mol,經驗證表明模型可靠。該研究旨在揭示氯化鉀和熱法磷酸制備磷酸二氫鉀的宏觀反應過程的相關規律,為設計反應器和優化工藝條件提供一定的理論依據。
磷酸二氫鉀;反應動力學;活化能
磷酸二氫鉀在農業、化工、醫藥、食品、飼料等領域都有廣泛的應用。因其具有磷鉀含量高、化學性質穩定、易溶于水等優點,在市場上倍受歡迎。其生產方法主要有中和法、傳統復分解法、萃取法、電解法等[1]。中和法[2-3]以KOH或K2CO3和H3PO4為原料,利用酸堿中和原理制取磷酸二氫鉀,具有工藝簡單、生產技術成熟等優點,缺點是原料成本高;傳統復分解法[4]以氯化鉀飽和溶液與熱法磷酸反應,反應溫度高,反應周期長,對設備的腐蝕嚴重;萃取法[5-6]存在萃取劑價格昂貴、萃取劑再生回收伴隨損失、失活現象等問題,導致原料成本較高;離子膜電解法[7-8]具有工藝流程短、產品純度高等特點,但其設備投資大,未實現工業化生產。筆者采用固體氯化鉀與熱法磷酸在通入蒸汽和負壓的條件下進行反應,可得到磷酸二氫鉀。該制備工藝反應條件溫和、對設備腐蝕較小、工藝簡單。該反應屬于液固非催化反應過程,其中氯化鉀與磷酸反應的速率決定了工藝實施難易程度。目前尚無關于該反應過程的宏觀動力學過程研究的相關文獻,因此筆者進行了該過程的反應動力學實驗研究,進一步描述氯化鉀與熱法磷酸反應的機理,為工業生產磷酸二氫鉀提供基礎數據和理論依據。
1 實驗部分
1.1 試劑與儀器
氯化鉀(分析純,粉末狀,質量分數≥99.8%);熱法磷酸(分析純,P2O5質量分數為60.58%)。
SHB-Ⅲ循環水多用真空泵;DF-101S集熱式恒溫加熱磁力攪拌器。
1.2 實驗原理
以氯化鉀和熱法磷酸為原料,在反應溫度為373~413 K、通入蒸汽和負壓條件下反應可制備出磷酸二氫鉀。根據化學反應平衡原理,增加反應物的濃度和降低生成物的濃度,移除體系中的氯化氫氣體可使平衡向右移動,使反應趨于完成。采用通蒸汽和抽真空的方式,不斷地使氯化氫氣體離開反應體系,使平衡向右移動。
其反應方程式如下:
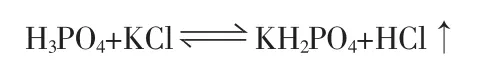
1.3 實驗方法
將氯化鉀與熱法磷酸加入500mL圓底燒瓶中,保持真空度在-0.09MPa,加熱物料并向其中通入蒸汽,保持溫度恒定,并攪拌。待反應完畢后,取樣測定反應產物中Cl-的含量,計算氯化鉀的轉化率。
氯化鉀轉化率的計算式為:
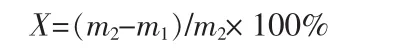
式中:X為氯化鉀轉化率,%;m1為反應后Cl的質量,g;m2為反應前Cl的質量,g。
實驗中采用GB/T 3051—2000無機化工產品中氯化物含量測定的通用方法——汞量法考察Cl-的含量。
2 實驗結果與討論
2.1 反應條件對氯化鉀轉化率的影響
圖1為不同溫度下反應時間與氯化鉀轉化率的關系曲線圖。由圖1可以看出,在不同溫度下,氯化鉀轉化率隨反應時間變化的規律相似。隨著反應溫度的升高,曲線上移,說明溫度升高,化學反應速率加快。提高溫度有利于氯化鉀與磷酸的復分解反應朝右進行。氯化鉀轉換率隨反應時間的延長而增加,表明該復分解反應受反應時間的影響較大。在0~ 3 h內,氯化鉀的轉化率隨反應時間延長而增加的速度較快;在3 h后,轉化率隨反應時間延長而增加的速度變慢,直至轉化率基本保持不變。其原因可能是隨著反應時間的延長,料漿黏稠度變大,出現物料顆粒表面包裹,自阻化現象加劇所致。

圖1 不同溫度下氯化鉀的轉化率與反應時間的關系
在反應時間為4 h、真空度為-0.09MPa的條件下,保持其他條件恒定,氯化鉀轉化率隨磷酸與氯化鉀物質的量比(簡稱原料配比)和反應溫度的變化曲線如圖2所示。由圖2可以看出,在不同原料配比時,氯化鉀轉化率隨反應溫度變化的趨勢基本相似。隨著原料配比的增加轉化率逐漸增加,說明增加磷酸的量,有利于反應朝右進行。同時氯化鉀轉化率隨反應溫度的升高而增加,在363~413K氯化鉀的轉化率隨反應溫度升高而增加的速度較快,在413 K后,轉化率增加的速度變慢,可見該反應受溫度的影響較大。
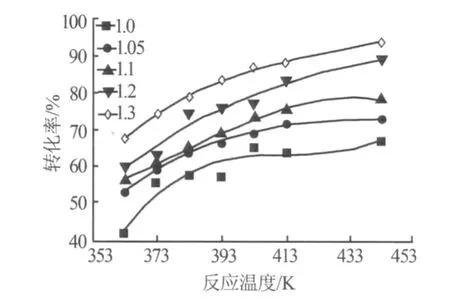
圖2 不同原料配比時氯化鉀轉化率隨反應溫度的變化
2.2 反應模型與參數的求解
流體與固體反應的經典理論模型有收縮未反應芯模型、整體反應模型、有限厚度反應區模型、微粒模型、單孔模型、破裂芯模型等[9]。目前,國內未見用數學模型來描述固體氯化鉀與磷酸反應的過程。考慮本實驗的實際情況:固體氯化鉀和磷酸反應,隨著反應時間的延長和反應溫度升高所表現出的明顯自阻化現象,可采用德羅茲多夫動力學方程描述動力學模型[10-11]。
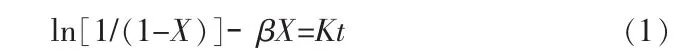
式中:X為氯化鉀的轉化率,%;t為反應時間,min;K為總反應速率常數,min-1;β為阻緩系數。
對不同溫度下的反應時間及對應的氯化鉀轉化率數據,用(1)式進行線性擬合,分別以1/t ln[1/(1-X)]對X/t作圖,如圖3所示。由直線得到相應的截距和斜率,截距即為總反應速率常數K,斜率為阻緩系數β,結果見表1。
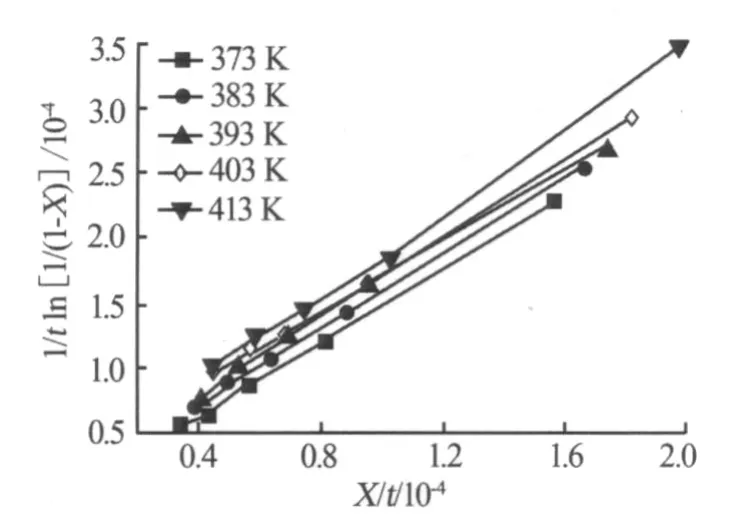
圖3 不同溫度下1/t ln[1/(1-X)]與X/t的關系
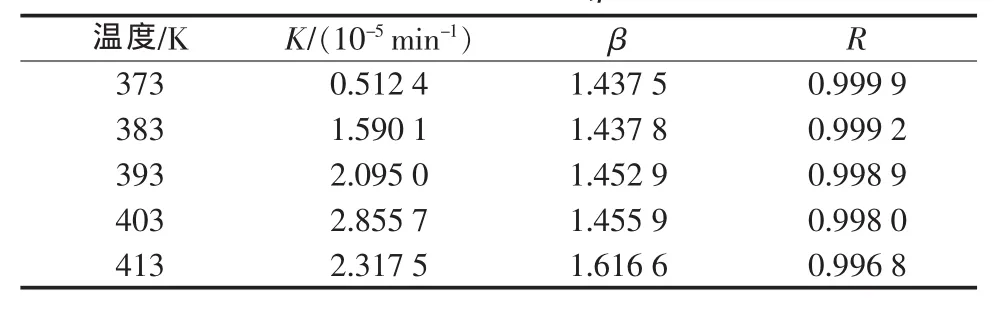
表1 不同溫度下K,β和R值
從圖3可以看出,采用式(1)擬合實驗數據,1/t ln[1/(1-X)]與X/t呈較好的線性關系。從表1可以看出,直線擬合的相關系數R均在0.99以上,表明1/t ln[1/(1-X)]與X/t的相關程度較高。總反應速率常數隨著溫度的升高而增大,溫度升高到413 K略降低。阻緩系數隨反應溫度的升高有增大的趨勢,在實驗溫度條件范圍內變化較大,且當溫度升至413 K阻緩系數突然變大,說明溫度過高反應料漿黏稠度增加,導致物料顆粒表面包裹,出現自阻化現象。由于受阻化作用的影響,反應速率減小。這可能就是轉化率變化先快后慢的原因。
2.3 反應表觀活化能的求取
總反應速度常數是溫度的函數,與溫度的關系服從Arrhenius公式[12],即:
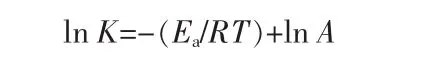
式中:A為指前因子,min-1;Ea為反應活化能,kJ/mol。
根據表1中的總反應速率常數K及Arrhenius公式,在373~413 K下做-ln K與1/T的關系曲線圖,見圖4。將圖4中的-ln K與1/T的關系曲線進行直線線性擬合,其相關系數R為0.961 1,表明-ln K與1/T的線性關系良好,符合Arrhenius公式。由圖4可得到截距為9.158 87,斜率為7 919.51。經計算可得磷酸與氯化鉀反應表觀活化能為65.84 kJ/mol,A=2.215 min-1;K與溫度的關系表達式為 K= exp(-65 840/RT)+2.215。因此,磷酸與氯化鉀反應的動力學模型方程為 1/t ln[1/(1-X)]-βX/t=exp(-65 840/RT)+2.215。
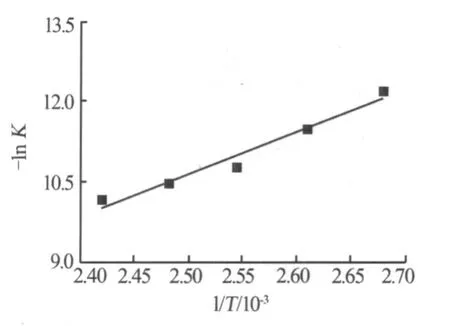
圖4 -ln K與1/T的關系
2.4 動力學方程的驗證
用反應溫度為403 K的實驗數據,以轉換率X為橫坐標,以時間t為縱坐標,作散點圖,見圖5。對散點按照式1/t ln[1/(1-X)]-βX/t=K進行線性擬合,其相關系數R為0.944 8。從圖5可以看出,德羅茲多夫動力學方程的模擬值與實驗數據很接近,相差不大。因此,該動力學模型方程的相關性良好,能夠較好地反映實驗結果,能較好地描述磷酸與氯化鉀的復分解反應。
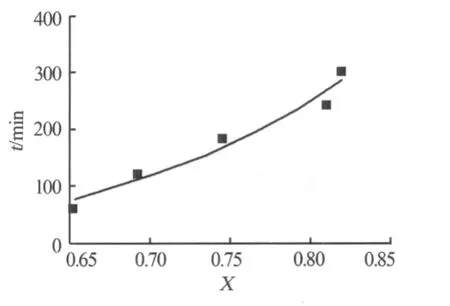
圖5 實驗值與模擬值的對比
3 結論
通過系統研究熱法磷酸與氯化鉀在反應溫度為373~413 K、n(H3PO4)∶n(KCl)=1.0~1.3條件下的反應動力學,結果表明:在不同原料配比時,氯化鉀轉化率隨反應溫度變化的規律基本相似,轉化率隨著原料配比的增加而逐漸增加;該反應過程可通過德羅茲多夫模型 1/t ln[1/(1-X)]-βX/t=exp(-65 840/ RT)+2.215來描述,在反應初期的0~3 h,氯化鉀的轉化率隨反應時間延長而增加的速度較快,受阻化作用的影響,在3 h后轉化率隨反應時間延長而增加的速度變慢;氯化鉀與磷酸復分解反應的表觀活化能為65.84 kJ/mol,阻化作用對反應速率的影響較大,阻緩系數也隨反應溫度升高而增大,要想進一步提高反應的轉化率可適當調節蒸汽的通入量,降低物料的黏稠度,或采用其他方法結合復分解法,以實現短時間內獲得較高的產品轉化率。
[1] 陳嘉甫,譚光薰.磷酸鹽的生產與應用[M].成都:成都科技大學出版社,1989:67-89.
[2] 陳朝銀,趙聲蘭,黨潔修,等.媒法由濕法磷酸生產純凈磷酸二氫鉀的研究[J].化工礦物與加工,1999,28(7):4-7.
[3] 李玲君,武沂蒙.一步結晶法制取磷酸二氫鉀的研究[J].河南化工,1999(8):18-19.
[4] 周建中,蘇元復.液液非均相復分解反應制取磷酸鹽的研究[J].化學世界,1985,26(1):2-3.
[5] 陳路萍,樊繼寬.濕法磷酸生產磷酸二氫鉀新工藝[J].淮海工學院學報:自然科學版,2001,10(2):39-41.
[6] 李海麗,曾波.萃取法生產磷酸二氫鉀工藝[J].云南化工,2001,28(1):14-16.
[7] Jean-Louis B,FrancoisP.Electrolytic process forobtaining chlorine in a pure stateand alkalimetalphosphates in concentrated solution and a cell for accomplishing this process:US,3763005[P].1973-10-02.
[8] Floyd Lester Ramp.Electrolytic cation exchange process for conjoint manufacture of chlorine and phosphate salts:US,3974047[P]. 1976-08-10.
[9] 朱炳辰.化學反應工程 [M].3版.北京:化學工業出版社,2004:345-352.
[10] 余靜.利用低品位磷礦生產濕法磷酸的新工藝及動力學研究[D].成都:四川大學,2005:48-50.
[11] 李成蓉,鐘本和,張允湘,等.金河磷礦在硫、磷混酸中的溶解動力學[J].化工學報,1998,49(3):336-341.
[12] 陳五平.無機化工工藝學 (三):化學肥料[M].2版.北京:化學工業出版社,1989:230.
Research on double decomposition reaction kineticsof potassium chlorideand phosphoric acid
Gou Ping,Fu Quanjun,Wang Xinlong,Yang Lin,Fu Yuxin,Zhang Zhiye
(SchoolofChemical Engineering,Sichuan University,Chengdu 610065,China)
Using potassium chlorideand thermalphosphoric acid as rawmaterials,the double decomposition reaction process to prepare potassium dihydrogen phosphate was studied.Influences of reaction time,reaction temperature,and mix ratio of materials on the conversion rate of potassium chloridewere investigated.Under the conditions of the reaction temperature at 373~413 K and the reaction time at1~4 h,themacroreaction processofpotassium chloride and thermalphosphoric acid was studied.Self-impedingeffectplayed an important roleon the reaction rate,and its coefficient(β)increasedwith the increasing temperature.Drozdov equation was selected to simulate the reaction data and the dynamic equationwas confirmed:1/t ln[1/(1-X)]-βX/t=exp(-65840/RT)+2.215.Apparentactivationenergy(Ea)was65.84 kJ/mol,and themodewas reliable.Studywas expected to reveal the relatedmechanism of the preparation for potassium dihydrogen phosphatewith potassium chloride and thermal phosphoric acid,so as to provide a necessary theoretical basis for the design of reactor and for optimization of processing conditions.
potassium dihydrogen phosphate;reaction kinetics;activation energy
TQ131.13
A
1006-4990(2012)04-0013-03
四川省科技支撐計劃(2008GZ0025)。
2011-10-12
茍蘋(1985— ),女,在讀碩士研究生,主要從事精細磷酸鹽工藝的研究。
聯 系 人:張志業
聯系方式:nic1201@163.com
猜你喜歡
作文·小學低年級(2025年2期)2025-02-13 00:00:00
小雪花·小學生快樂作文(2024年11期)2024-12-31 00:00:00
作文·小學低年級(2024年2期)2024-04-29 00:00:00
作文·小學低年級(2023年3期)2023-04-29 00:00:00
小獼猴智力畫刊(2022年9期)2022-11-04 02:31:42
小主人報(2022年4期)2022-08-09 08:52:06
中學生數理化·中考版(2022年11期)2022-02-16 07:01:20
小哥白尼(趣味科學)(2019年6期)2019-10-10 01:01:50
發明與創新(2016年38期)2016-08-22 03:02:52
太空探索(2016年5期)2016-07-12 15:17:55
