HPLC法測定頸腰康片中士的寧的含量
2013-03-06 07:57:16李文君
中國中醫藥現代遠程教育 2013年6期
張 進 李文君
(吉林省通化市食品藥品檢驗所中藥室,通化134001)
HPLC法測定頸腰康片中士的寧的含量
張 進 李文君
(吉林省通化市食品藥品檢驗所中藥室,通化134001)
目的 建立以HPLC法檢測頸腰康片中士的寧的含量測定。方法 色譜柱:依利特C18(4.6×250mm,5μm);流動相:乙腈-0.01mol/L庚烷磺酸鈉與0.02mol/L磷酸二氫鉀等量混合溶液(用10%磷酸調節pH值2.8)(23∶77);流速:1.0ml/min;柱溫:室溫;檢測波長:254nm;以士的寧為對照品,采用外標法定量。結果 士的寧線性范圍為0.0247-0.2223μg(r=1.0000),平均回收率為99.80%,RSD為1.29%。結論 本法簡便,結果準確,重現性好,可用于該藥品的質量控制。
頸腰康片;士的寧;HPLC;含量測定
頸腰康片是由制馬錢子、伸筋草、香加皮、乳香(醋炒)、沒藥(醋炒)、紅花等十味中藥組成,其中制馬錢子為本處方中的君藥之一。制馬錢子為馬錢科植物馬錢Strychnos nux-vomica L的干燥成熟種子的炮制加工品,主要成份為番木鱉堿(士的寧)、馬錢子堿、偽番木鱉堿、番木鱉次堿等,均屬生物堿類成分。具有通絡止痛、散結消腫等作用,用于風濕頑痹,麻木癱瘓、跌撲損傷、類風濕性關節痛等癥。為了有效地控制頸腰康片的內在質量,我們以制馬錢子中士的寧作為指標性成分,采用反相高效液相色譜法測定制馬錢子中士的寧的含量,方法易于操作,結果準確,重現性好專屬性強,現報告如下。
1 儀器與試藥
日本島津LC-2010AHT高效液相色譜儀,士的寧對照品(由中國藥品生物制品檢驗所提供)批號:110715-200212;規格:供含量測定用。乙腈為色譜純,水為純化水,其它試劑均為分析純。
2 方法與結果
2.1 色譜條件 色譜柱:依利特C18(4.6×200mm,5μm);流動相:乙腈-0.01mol/L庚烷磺酸鈉與0.02mol/L磷酸二氫鉀等量混合溶液(用10%磷酸調節p H值2.8)(23∶77);流速:1.0ml/min;柱溫:室溫;檢測波長:254nm;理論板數按士的寧峰計算應不低于6000。
2.2 溶液的制備
2.2.1 對照品溶液的制備 精密稱取士的寧對照品12.5mg置50ml量瓶中,加三氯甲烷至刻度,再精密吸取1ml置10ml容量瓶中,加甲醇稀釋至刻度,即得。
2.2.2 供試品溶液的制備 取本品10片,除去包衣,精密稱定,研細,精密稱取約1.2g,置具塞錐形瓶中,精密加入氫氧化鈉試液3ml,放置30分鐘,再精密加入三氯甲烷20ml,稱定重量,加熱回流2小時,放冷,再稱定重量,用三氯甲烷補足減失的重量,搖勻,分取三氯甲烷提取液,用鋪有少量無水硫酸鈉的濾紙濾過,棄去初濾液,精密量取續濾液3ml,置10ml量瓶中,用甲醇稀釋至刻度,搖勻,即得。
2.2.3 陰性對照溶液的制備 按處方比例配成不含制馬錢苷的陰性對照樣品,照供試品溶液的制備方法,制成陰性對照溶液。分別精密吸取陰性對照溶液、供試品溶液及對照品溶液各5μl,注入液相色譜儀中,按液相色譜條件測定,結果表明,陰性對照溶液色譜圖中與對照品相同保留時間處無干擾。
2.3 線性關系考察 精密稱取士的寧對照品適量加甲醇制成每1ml含25μg的溶液,分別精密吸取1、3、5、7、9μl測定,記錄色譜圖并測定峰面積,以峰面積(A)對進樣量(μg)回歸,回歸方程為:Y=2510579x-794(r =1.0000),結果表明士的寧在0.0247-0.2223μg范圍內,與峰面積呈良好的線性關系。
2.4 精密度試驗 精密吸取對照品溶液5μl,注入液相色譜儀,重復進樣5次,RSD為1.09%。
2.5 穩定性試驗 取同一批樣品的供試品溶液,分別于0、2、4、8、12h,測定含量,RSD為0.13%。結果表明供試品溶液在12h內穩定。
2.6 重復性試驗 取同一批樣品,按2.2.2項下操作方法獨立測定五次,用外標法計算士的寧含量(mg/片),分別為0.532、0.534、0.534、0.530、0.533;RSD為0.31%。
2.7 回收率試驗 精密稱取已知含量的樣品適量,精密加入一定量的對照品,按2.2.2項下操作,作為試驗溶液;分別精密量取對照品溶液和試驗溶液各5μl進樣,計算回收率(結果見表1)。平均回收率99.80%,RSD= 1.29%,結果表明本法回收率較好,準確度較高。
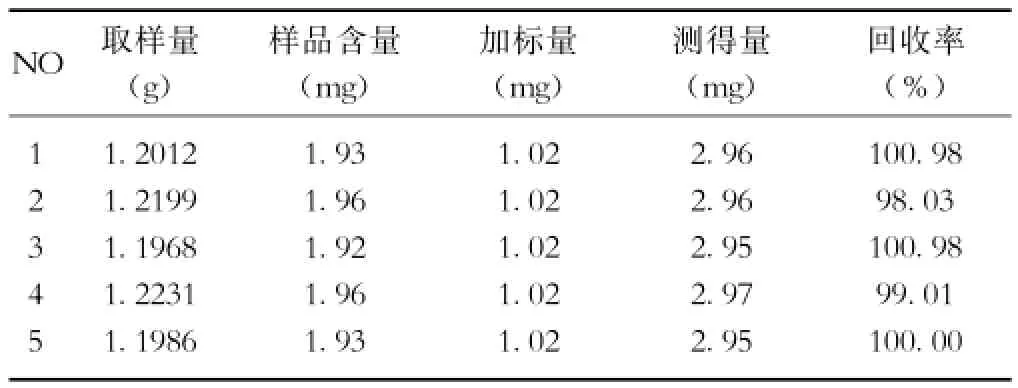
表1 回收率試驗結果
2.8 樣品測定 取不同批號的頸腰康片,按2.2.2項下操作,分別精密量取對照品溶液和樣品溶液各5?l,注入高效液相色譜儀,測定士的寧的含量(結果見表2)。

表2 三批樣品中士的寧的含量測定結果
3 討論
3.1 檢測波長的選擇 取士的寧對照品的甲醇溶液,經UV-260紫外分光光度計200nm~400nm范圍內測定,結果士的寧在254nm波長處,有最大吸收故選擇254nm為本實驗的測定波長。
3.2 流動相的選擇 參照有關資料[1]我們對乙腈-0.01mol/L庚烷磺酸鈉與0.02mol/L磷酸二氫鉀等量混合溶液(用10%磷酸調節pH值2.8)(21∶79)、乙腈-0.01mol/L庚烷磺酸鈉與0.02mol/L磷酸二氫鉀等量混合溶液(用10%磷酸調節PH值2.8)(22:78)、乙腈-0.01mol/L庚烷磺酸鈉與0.02mol/L磷酸二氫鉀等量混合溶液(用10%磷酸調節p H值2.8)(23∶77)進行優化試驗,結果,以乙腈-0.01mol/L庚烷磺酸鈉與0.02mol/L磷酸二氫鉀等量混合溶液(用10%磷酸調節p H值2.8)(23∶77)為流動相分離效果較好,保留時間適度,因此,確定其為本法的流動相。
3.3 樣品提取方法的考察 我們采用了三氯甲烷50ml與濃氨試液1ml,密塞,輕輕振搖,稱定重量,放置24小時及精密加入氫氧化鈉試液3ml,放置30分鐘,再精密加入三氯甲烷20ml,稱定重量,加熱回流2小時。結果后者提取方法即節省時間又提取充分,因此我們采用正文所述的供試品溶液的制備方法。
[1]國家藥典委員會.中華人民共和國藥典(一部)[M].北京:化學工業出版社,2010.
10.3969/j.issn.1672-2779.2013.06.113
1672-2779(2013)-06-0157-02
??韓世輝
2013-03-12)
