核磁共振內標法測定蘇丹紅I對照品的含量
2013-04-23 01:28:52耿巖玲馬然劉偉高紅梅劉建華王曉段文娟
山東科學 2013年5期
耿巖玲,馬然,劉偉,高紅梅,劉建華,王曉,段文娟*
(1.山東省科學院中藥過程控制研究中心,山東省分析測試中心,山東 濟南 250014;2.山東師范大學生命科學學院,山東 濟南 250014)
蘇丹紅是一種人工合成的紅色染料,常作為工業染料廣泛應用于石油、機油和其他的一些工業溶劑中,目的是增色,也可用于鞋、地板等的增光。蘇丹紅有Ⅰ、Ⅱ、Ⅲ、Ⅳ號4種,該類物質具有偶氮結構,毒理學研究表明其具有致突變性和致癌性,在我國和世界上許多國家都禁止用于食品中。但由于具有良好的光澤持久性與穩定性,不法商販將其用于改善食品的感官色澤。近年來,蘇丹紅色素,特別是蘇丹紅I,經常被添加到辣椒及相關食品、熟食和口紅中,對人類健康的危害非常大[1-2]。目前蘇丹紅I的含量測定方法主要有液相色譜法、薄層色譜法、色質聯用法等[3-5],雖然采用色譜法進行含量測定在分離上具有一定的優勢,但是由于殘留的溶劑、無機物等雜質與被測物質色譜響應值不同,造成了樣品的絕對含量測定結果不準確。而且,采用以上方法測定含量均需要對照品。
早在20世紀70年代,就有關于核磁共振定量分析的報道。隨著超導高磁場的脈沖Fourier變換核磁共振譜儀的應用,核磁共振定量分析方法在準確度、靈敏度及分析速度等方面都得到了極大的提高,已經接近HPLC的水平,而且這種分析法具有所需樣品量少、掃描時間短以及無需標準品等優點[6]。核磁共振定量分析法用于樣品的含量測定已有文獻報道,美、英等國和地區的藥典中也先后引入了此方法[7-10],但是有關采用此方法進行蘇丹紅含量測定的研究未見報道。本研究采用核磁共振內標法對蘇丹紅I對照品的含量進行測定,以期建立在沒有對照品的情況下蘇丹紅I的含量測定和質量控制方法,并為其他化學對照品含量的測定方法提供參考。
1 實驗部分
1.1 儀器
Varin Inova 600mHz超導核磁共振波譜儀(美國 Varian公司)。
1.2 試劑
氘代氯仿 (美國CIL公司,氘代度>99.9%),苯甲酸(美國Sigma公司,批號101253928,純度99.2%),蘇丹紅I對照品(山東省分析測試中心研制)。蘇丹紅I和苯甲酸的結構見圖1。

圖1 蘇丹紅I和苯甲酸的化學結構式Fig.1 Chemical structure of Sudan I and benzoic acid
1.3 實驗條件
測定溫度:25 ℃,FT size:131072,譜寬:15000.9 Hz,環境濕度:35%,觀察頻率:599.773 MHz,采集時間:3.000 s,累加次數:32。
1.4 實驗方法
根據文獻報道[11],分別精密稱取適量蘇丹紅I對照品和苯甲酸對照品,置于直徑5 mm的核磁共振樣品管中,加適量氘代氯仿試劑振蕩溶解,制成待測試樣溶液。在上述實驗條件下調整儀器參數,勻場、采樣,得到圖譜后再進行相位和基線調整,對蘇丹紅I和苯甲酸的定量峰分別進行積分,按照1H-NMR內標法根據積分結果用以下公式計算樣品的質量分數。
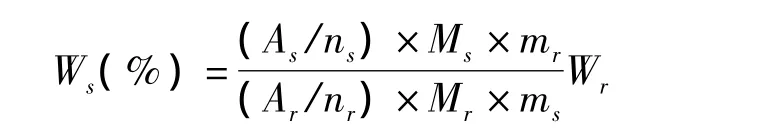
上述公式中,As:被測樣品定量峰的積分面積,ns:被測樣品定量峰包含的質子數,Ms:被測樣品的分子質量,Ar:內標物質定量峰的積分面積,nr:內標物質定量峰包含的質子數,Mr:內標物質的分子質量,mr:稱取的內標質量,Wr:內標的質量分數,ms:樣品的質量。
2 結果與討論
2.1 溶劑、內標和定量特征峰的選擇
測試溶劑應選擇對被測樣品和內標物溶解性都非常好且不造成干擾的氘代試劑。本研究選擇氘代氯仿為測試溶劑,該溶劑對蘇丹紅I和苯甲酸的溶解性都很好,且核磁共振氫譜上的溶劑峰在δ 7.26,對其他信號峰無干擾,不會影響含量測定結果。選擇苯甲酸為內標物,從苯甲酸的核磁共振氫譜中可以看到苯甲酸的2位和6質子信號在δ 8.13,與其他信號完全分開,因此,可作為內標物。
在定量分析之前,已經通過核磁共振一維譜和二維譜對目標化合物的信號進行了全歸屬。在蘇丹紅I的核磁共振氫譜上可以看到,5位的質子信號化學位移在δ 8.51,與其他信號完全分開,因此,可作為定量分析的特征峰。核磁共振氫譜法測定蘇丹紅I含量的圖譜見圖2。
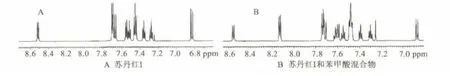
圖2 核磁共振氫譜法測定蘇丹紅I含量的圖譜Fig.2 1H-NMR spectrum of sudan I content determination
2.2 線性關系
按1.4的實驗方法制備6個不同濃度的樣品溶液,測定1H-NMR譜,分別對蘇丹紅I和苯甲酸特征信號峰的峰面積進行積分,每個樣品的積分面積取3次積分的平均值,結果見表1。
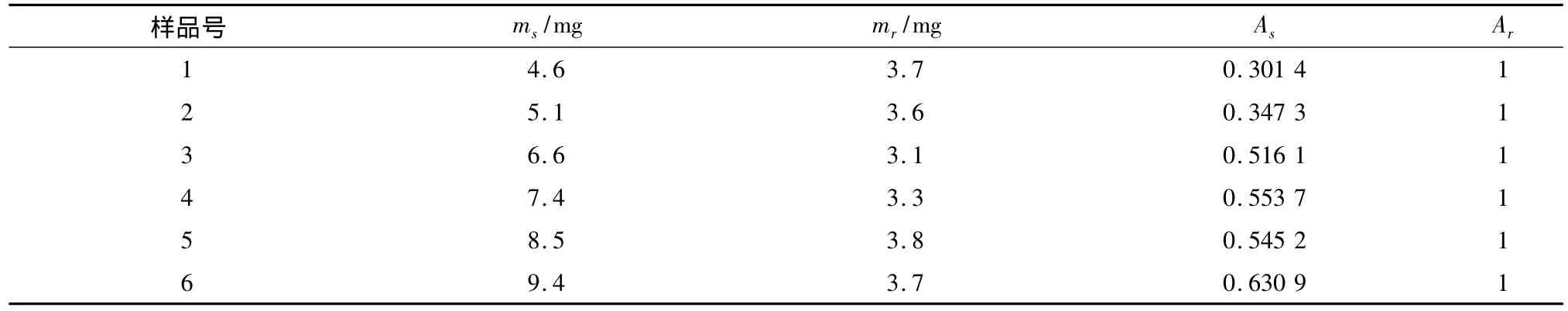
表1 蘇丹紅I與苯甲酸的質量及峰面積積分值Table 1 Mass and integral value of peak area of Sudan I and benzoic acid
以蘇丹紅I和苯甲酸的積分面積比值y對蘇丹紅I和苯甲酸質量比值x做線性關系圖,得回歸方程為y=0.2424x-0.0028,相關系數r=0.9982。結果表明,采用核磁共振內標法對蘇丹紅I的含量進行測定,線性關系良好。
2.3 重復性實驗
取2.2的2號樣品,在上述同一實驗條件下重復測定6次,以蘇丹紅I和苯甲酸定量特征峰的積分面積比值計算,檢測重復性,RSD值為0.53%,結果表明,此方法重復性良好。
2.4 穩定性實驗
取2.2的 2號樣品,室溫下放置,按1.4的實驗方法,分別于 0、4、6、8、10 h后測定,檢測穩定性,RSD 值為0.656%,實驗結果表明,樣品溶液在10 h內穩定。
2.5 加樣回收率實驗
分別取6份已知含量的蘇丹紅I樣品約5 mg,精密稱定,分別加入高、中、低3個濃度的蘇丹紅I標準品,按1.4的實驗方法測定1H-NMR譜,按回收率(%)=[測得含量-樣品含量]/標準品添加,計算回收率,所得結果見表2。
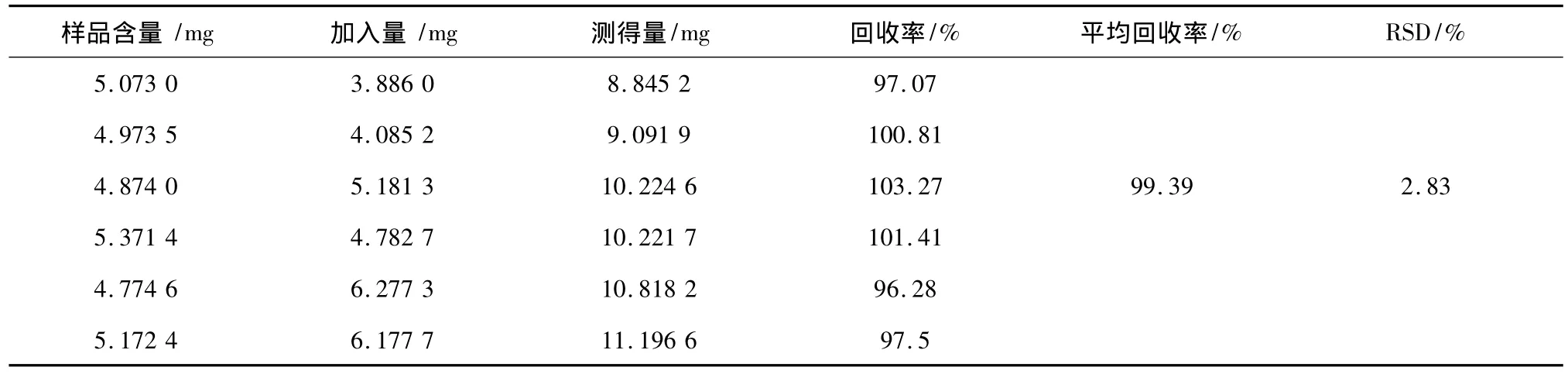
表2 加樣回收率實驗(n=6)Table 2 Experiment of the average recovery rate(n=6)
2.6 樣品測定
按照上述實驗條件和方法,測定3批蘇丹紅I對照品和苯甲酸特征峰的峰面積,根據1.4的計算公式計算出蘇丹紅I對照品的絕對質量分數,同時采用高效液相色譜(HPLC)法對此3批蘇丹紅I對照品的相對質量分數進行測定,結果見表3。由表3中可以看到,采用核磁共振波譜法測得蘇丹紅I對照品的質量分數略低于HPLC峰面積歸一化法。

表3 3批蘇丹紅I對照品含量測定結果(質量分數/%)Table 3 Determination result of the content of 3 batches of Sudan I
2.7 掃描次數的選擇
將掃描次數設定為4、8、16、32、64、128,對樣品分別進行實驗。結果表明,掃描次數越多,信號信噪比越好,但是掃描次數的改變對樣品和內標物積分面積的比值沒有明顯的影響,因此選擇掃描次數為32。
3 結論
實驗結果表明,采用核磁共振波譜法對蘇丹紅I含量進行測定,線性關系、重復性和穩定性均良好。此方法具有樣品量小、高效、簡便、無需對照品等優點,含量測定和結構鑒定可同步完成,可以用于其他化學對照品含量的測定。
[1]秦菲.食品中蘇丹紅的毒性及檢測方法[J].北京聯合大學學報:自然科學版,2008,22(2):50-54.
[2]張滿芳,王向紅,黃桂湘,等.蘇丹紅色素概述及蘇丹紅色素的檢驗[J].使用預防醫學,2008,15(4):1251-1252.
[3]洪祥奇,張揚.薄層色譜測定食品中蘇丹紅染料方法的探討[J].中國衛生檢驗雜志,2006,16(11):330-332.
[4]GB/T 19681-2005,食品中蘇丹紅染料的檢測方法高效液相色譜法[S].
[5]CALBIANI F,GARERI M,ELCIRI L,et al.Accurate mass measurements for the confirmation of Sudan azo-dyes in hot chilli products by capillary liquid chromatography-electro-spray tandem quadrupole orthogonal-acceleration time of flight mass spectrometry[J].J Chromatogr A,2004,1058(1/2):127-135.
[6]張友杰,劉小鵬.藥物核磁共振定量分析參數的研究[J].波譜學雜志,2007,24(3):289-295.
[7]于小波,相秉任,王國華,等.核磁共振法測定硫酸依替米星含量[J].中國抗生素雜志,2011,36(8):610-612.
[8]United States Pharmacopoeia Commission.United States Pharmacopoeia[M].US:US Pharmacopoeia Convention,Inc.,2000:1221-1222.
[9]楊金霞,姚婷玉,王建偉,等.核磁共振法測定西洛他唑含量[J].化學分析計量,2012,21(4):68-70.
[10]易進海,劉云華,陳燕,等.核磁共振波譜法測定蒿本內酯對照品的含量[J].藥物分析雜志,2010,30(4):680-682.
[11]蔣文,蘇敏,陳雙全,等.核磁共振氫譜內標法測定茚地普隆的含量[J].分析化學研究簡報,2008,36(3):385-388.
猜你喜歡
小獼猴智力畫刊(2022年9期)2022-11-04 02:31:42
中學生數理化·八年級物理人教版(2021年12期)2021-12-31 03:23:08
中學生數理化·中考版(2020年10期)2020-11-27 01:59:48
小哥白尼(趣味科學)(2019年6期)2019-10-10 01:01:50
中國生殖健康(2019年2期)2019-08-23 08:12:08
發明與創新(2016年38期)2016-08-22 03:02:52
太空探索(2016年5期)2016-07-12 15:17:55
Coco薇(2016年2期)2016-03-22 02:42:52
汽車觀察(2016年3期)2016-02-28 13:16:26
Coco薇(2015年1期)2015-08-13 02:47:34
