壞死性凋亡參與NMDA誘導大鼠皮層神經元的興奮性神經毒作用
2013-09-13 09:06:30李艷麗
中西醫結合心腦血管病雜志 2013年2期
李艷麗,張 策,張 虹
谷氨酸是中樞神經系統重要的興奮性神經遞質。谷氨酸受體的活化在突觸可塑性、學習和記憶中起重要作用[1]。在中樞神經系統,興奮性突觸傳遞主要由離子型谷氨酸受體介導。離子型谷氨酸受體包括AMPA受體、KA受體和NMDA受體[2]。
NMDA受體為配體門控離子通道,在許多突觸活動中起重要作用[3]。但是NMDA受體的過度活化,可引起細胞內鈣超載,引發一系列酶的級聯反應,并最終誘導神經元死亡,被稱為“興奮性神經毒”[4]。許多急性或慢性腦組織損傷性疾病都與NMDA受體介導的興奮毒性有著重要的聯系,比如中風、癲癇和某些神經變性疾病等[5]。數據顯示NMDA型谷氨酸受體通道在創傷性腦損傷的病理生理過程中具有重要的作用[6]。而且NMDA受體的過度活化是創傷性腦損傷引起神經學缺陷的主要原因,NMDA受體拮抗劑可抑制這些癥狀[7]。
傳統上,細胞死亡被分為細胞壞死和凋亡。凋亡是一種程序化的主動性細胞死亡,且無炎癥反應。壞死是病理產生的被動死亡,且這種細胞死亡方式會導致炎癥物質的釋放[8,9]。最近,Degterev等[10]研究發現,在細胞內不存在凋亡信號時,Fas/TNFR能活化一種非凋亡的死亡途徑,稱為壞死性凋亡。因同時具有壞死和凋亡的生化特征和超微結構特征而得名。同時。他們發現一種小分子物質 Necrostatin-1(Nec-1),可以特異且有效地抑制壞死性凋亡,為壞死性凋亡的鑒別提供了一個有效的工具。迄今為止,壞死性凋亡的機制并不清楚,Nec-1的發現為評價壞死性凋亡的特征及作用提供了唯一的可能。因此,本研究通過觀察Nec-1對NMDA誘導的興奮毒的抑制作用,從而明確NMDA誘導的興奮神經毒中是否存在壞死性凋亡。
1 材料與方法
1.1 試 劑 DMEM/F-12 和 胎 牛 血 清 (Gibco-BRL);Cell Counting Kit-8(CCK-8)、calcein-AM(Dojindo);NMDA、阿糖胞苷、MK-801、多聚賴氨酸、Nec-1(Sigma);胰蛋白酶(Biosharp);乳酸脫氫酶(LDH)(南京建成生物工程研究所)。
1.2 原代細胞培養 取新生1d~3dWistar大鼠,分離大腦皮層,用胰酶消化制備單細胞懸液。細胞懸液經1 000r/min離心5min,棄上清,用完全培養液(DMEM/F-12培養基+15%胎牛血清)重懸細胞。然后接種于培養瓶或培養板上,在37℃,5%CO2的孵育箱培養。24h后加入10μmol/L阿糖胞苷。成熟的神經元(8d~10d)用于實驗。
1.3 NMDA誘導興奮性神經毒 將成熟神經元暴露于NMDA。用無鎂Locke’s液洗滌細胞兩次,然后用無鎂Locke’s液(含或不含NMDA)孵育細胞。2h后將毒性介質移出,相應的培養液返回[11]。Nec-1組在毒性誘導前24h加入不同濃度的Nec-1(10μmol/L,30μmol/L,100μmol/L)預孵育。
1.4 細胞活力檢測 細胞在96孔板培養(每孔100μL),NMDA處理2h后,將毒性介質移出,培養液返回。繼續培養24h,向各孔加入10μL CCK-8溶液,在37℃,5%CO2的孵育箱孵育2h,用酶標儀測定490nm處OD值。
1.5 LDH檢測 當細胞遭受損傷時,LDH由胞漿釋放到胞外。NMDA毒性誘導之后,繼續培養24h,收集培養液檢測LDH活性。
1.6 calcein-AM染色 在Locke’s液介質中細胞與calcein-AM避光孵育20min,PBS避光洗滌兩次,在490nm激發波長,515nm發射波長的濾光片的熒光顯微鏡下觀察細胞。
2 結 果
2.1 Nec-1抑制NMDA引起的細胞活力的下降 CCK-8細胞活力檢測發現NMDA引起皮層神經元興奮毒損傷(見圖1)。與對照組相比,將神經元暴露于NMDA(100μmol/L)2h使細胞活力降至(42.55±2.86)%(P<0.01)。NMDA引起的神經毒可以被NMDA受體的非競爭性拮抗劑 MK-801(10μmol/L)完全阻斷,說明這種損傷是由NMDA引起的。
在NMDA作用前24h用Nec-1預處理神經元可以抑制NMDA引起的細胞活力的下降,也就是說Nec-1預處理引起細胞 活 力 的 增 加 (見 圖 1)。Nec-1(30μmol/L)和 Nec-1(100 μmol/L)分別使其增加12%(P<0.05)和26%(P<0.01),但Nec-1(10μmol/L)并不能抑制NMDA引起的細胞活力的降低。
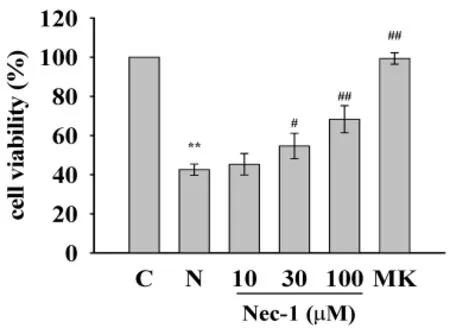
圖1 Nec-1抑制NMDA引起的細胞死亡
2.2 Nec-1抑制NMDA引起的LDH釋放的增加 皮層神經元暴露于NMDA可引起LDH的釋放(見圖2)。NMDA處理后,LDH水平與對照組相比增加54.38%(P<0.01),MK-801(10μmol/L)完全阻斷 NMDA引起的 LDH 的釋放。Nec-1(30 μmol/L)和 Nec-1(100μmol/L)預處理分別使 LDH 釋放減少14%(P<0.05)和28%(P<0.01)。
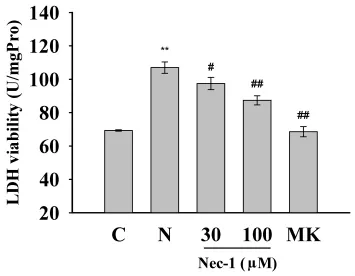
圖2 Nec-1抑制NMDA引起的LDH釋放
2.3 Nec-1抑制NMDA引起的活細胞數的減少 calcein-AM染色結果發現NMDA可引起活細胞數減少。NMDA處理后,calcein-AM染色陽性的神經元(代表活細胞)與對照組相比減少55.87% (P<0.01)。 與 NMDA 處 理 組 相 比,Nec-1(30 μmol/L)和Nec-1(100μmol/L)分別使活細胞數增加11%(P<0.05)和23%(P<0.01)。
3 討 論
谷氨酸是中樞神經系統重要的興奮性神經遞質。谷氨酸受體的活化在生理狀態下起重要作用[1]。同時,谷氨酸也可產生毒性作用,引起NMDA受體的過度活化,被稱為興奮性神經毒[12]。越來越多的研究表明,在許多急慢性神經性疾病,興奮性神經毒是引起神經元死亡的機制之一,包括亨廷頓癥(Huntington,s disease,HD)[13]、帕金森 氏癥(Parkinson’s disease,PD)[14]和阿爾茨海默病(Alzheimer’s disease,AD)[15]。在腦缺血或中風疾病中,興奮性神經毒也是引起神經元壞死或凋亡的機制。
壞死性凋亡是新近發現的一種細胞死亡方式,其特征為壞死的細胞死亡形態和自噬的活化[10]。據報道Nec-1可選擇性抑制壞死性凋亡,用來鑒定壞死性凋亡的存在。研究發現大腦中動脈阻塞后,腦室內注射Nec-1可顯著減少梗死面積,表明腦缺血損傷中有壞死性凋亡的發生[10]。
本研究最重要的發現是Nec-1抑制NMDA誘導的培養皮層神經元的損傷。已證實Nec-1抑制NMDA引起的細胞活力的降低、LDH釋放和活細胞數的減少。這些結果均表明壞死性凋亡參與了NMDA誘導的細胞損傷,也就是說在NMDA誘導的興奮性神經毒有壞死性凋亡的發生。
眾所周知,NMDA受體過度活化是引起腦缺血神經元死亡的主要原因[16]。NMDA受體過度活化在許多急慢性腦損傷中起重要作用,如中風或腦缺血及某些神經變性疾病[5]。在這些神經病理過程中,由于起始損傷的嚴重程度不同,NMDA可誘導神經元發生凋亡或者壞死[8]。本研究證實NMDA可誘導壞死性凋亡,為系統地理解與興奮性神經毒有關的分子途徑提供了新的證據。以前的研究表明在缺血性腦損傷中存在壞死性凋亡[10],本研究表明腦缺血中存在的壞死性凋亡可能是由于缺血引起谷氨酸的過量釋放引起NMDA受體的過度活化所致。
總之,NMDA誘導的興奮性神經毒包括多種細胞死亡方式:壞死、凋亡和壞死性凋亡。壞死性凋亡在其中起重要作用,為興奮性神經毒有關疾病的治療提供了新的靶點。而且壞死性凋亡級聯反應是可調節的,所以它可能會提供一個新的甚至更有效的治療靶點。
[1]Maragakis NJ,Rothstein JD.Glutamate transporters in neurologic disease[J].Arch Neurol,2001,58(3):365-370.
[2]Gasic GP,Hollmann M.Molecular neurobiology of glutamate receptors[J].Annu Rev Physiol,1992,54:507-536.
[3]Liu Y,Zhang J.Recent development in NMDA receptors[J].Chin Med J(Engl),2000,113(10):948-956.
[4]Takei N,Endo Y.Ca2+ionophore-induced apoptosis on cultured embryonic rat cortical neurons[J].Brain Res,1994,652(1):65-70.
[5]Choi DW.Glutamate neurotoxicity and diseases of the nervous system[J].Neuron,1988,1:623-634.
[6]Han RZ,Hu JJ,Weng YC,etal.NMDA receptor antagonist MK-801reduces neuronal damage and preserves learning and memory in a rat model of traumatic brain injury[J].Neurosci Bull,2009,25(6):367-375.
[7]Hayes RL,Jenkins LW,Lyeth BG,etal.Pretreatment with phencyclidine,an N-methyl-D-asparatate antagonist,attenuates long-term behavioral deficits in the rat produced by traumatic brain injury[J].J Neurotrauma,1988,5(4):259-274.
[8]Fink SL,Cookson BT.Apoptosis,pyroptosis,and necrosis:Mechanistic description of dead and dying eukaryotic cells[J].Infect Immun,2005,73(4):1907-1916.
[9]Bonfoco E,Krainc D,Ankarcrona M,etal.Apoptosis and necrosis:Two distinct events induced,respectively,by mild and intense insults with N-methyl-D-aspartate or nitric oxide/superoxide in cortical cell cultures[J].Proc Natl Acad Sci USA,1995,92(16):7162-7166.
[10]Degterev A,Huang Z,Boyce M,etal.Chemical inhibitor of nonapoptotic cell death with therapeutic potential for ischemic brain injury[J].Nat Chem Biol,2005,1(2):112-119.
[11]Cebere A,Liljequist S.Ethanol differentially inhibits homoquinolinic acid-and NMDA-induced neurotoxicity in primary cultures of cerebellar granule cells[J].Neurochem Res,2003,28(8):1193-1199.
[12]Beal MF.Role of excitotoxicity in human neurological disease[J].Curr Opin Neurobiol,1992,2(5):657-662.
[13]Tabrizi SJ,Cleeter MW,Xuereb J,etal.Biochemical abnormalities and excitotoxicity in Huntington’s disease brain[J].Ann Neurol,1999,45(1):25-32.
[14]Beal MF.Excitotoxicity and nitric oxide in Parkinson’s disease pathogenesis[J].Ann Neurol,1998,44(3Suppl 1):S110-S114.
[15]Hynd MR,Scott HL,Dodd PR.Glutamate-mediated excitotoxicity and neurodegeneration in Alzheimer’s disease[J].Neurochem Int,2004,45(5):583-595.
[16]Bonde C,Noraberg J,Noer H,etal.Ionotropic glutamate receptors and glutamate transporters are involved in necrotic neuronal cell death induced by oxygen-glucose deprivation of hippocampal slice cultures[J].Neuroscience,2005,136(3):779-794.
