超高效液相色譜-串聯四級桿質譜法檢測鎮靜安神類中藥制劑及保健品中非法添加的9種化學藥品
2013-09-14 07:07:48張繼春儲忠英包綜方
中國藥業 2013年6期
關鍵詞:檢測
田 蘭,張繼春,陳 睿,儲忠英,包綜方
(上海市松江食品藥品檢驗所,上海 201600)
目前,失眠的人群越來越多,有一部分人不得不借助鎮靜安神類藥物維持正常的睡眠和生活。多數人認為中藥和保健品作用較緩和、不良反應小,更喜歡使用中藥制劑和保健品,但個別不法商家為牟取暴利,在中藥制劑及保健品中非法添加鎮靜催眠類化學藥,以增強藥效,這不僅使中成藥原有的藥效可能發生改變,同時也使患者的用藥安全得不到應有的保障[1]。此類藥物也不可避免地存在某些副作用,如出現困倦、嗜睡、乏力、頭暈,大劑量使用時可引起共濟失調、暫時性的遺忘和意識障礙,嚴重時還可能導致昏迷、呼吸抑制。此外,安眠藥還具有易產生耐受性和依賴性的特點[2]。況且,該類藥物的半衰期很長,達50 h。本試驗中選擇了氯硝西泮、氯氮、艾司唑侖、阿普唑侖、地西泮、硝西泮、三唑侖、奧沙西泮和馬來酸咪達唑侖9種易被非法添加的藥物為檢測對象。目前根據國家食品藥品監督管理局藥品檢驗補充檢驗方法和檢驗項目批準件匯編有關規定測定這9種化學藥物的方法有薄層色譜法、高效液相色譜-串聯四級桿質譜(HPLC-MS/MS)法分別測定[3]。筆者建立了同時測定非法添加9種化學藥品的超高效液相色譜-串聯四級桿質譜(UPLC-MS/MS)法,為強有力打擊制假售假違法者提供技術保障和科學依據。
1 儀器與試藥
Waters ACQUITY UPLC Quattro MicroTM超高效液相色譜串聯三重四級桿質譜聯用儀,MassLynx4.1質譜工作站。對照品氯硝西泮(批號為 171227-200302)、氯氮(批號為 171248-200301)、艾司唑侖(批號為 1219-0102)、阿普唑侖(批號為 171218-200603)、地西泮(批號為 171225-200302)、硝西泮(批號為171217-200402)、三唑侖(批號為 1230-9701)、奧沙西泮(批號為 171229-200603)和馬來酸咪達唑侖(批號為 171250-201002,含量為99.9%)均由中國藥品生物制品檢定所提供;樣品由藥監分局監督抽驗;甲醇為色譜純(購自Merk公司),甲酸選用進口色譜純,水為超純水。
2 方法與結果
2.1 檢測條件
色譜條件:Acquity UPLC超高效液相色譜系統,Acquity BEH C18柱(2.1 mm × 50 mm,1.7 μm);流動相 A 為甲醇,流動相B為0.1%甲酸溶液,按表1進行梯度洗脫;流速0.3 mL/min,進樣量 5 μL,柱溫為 35 ℃。
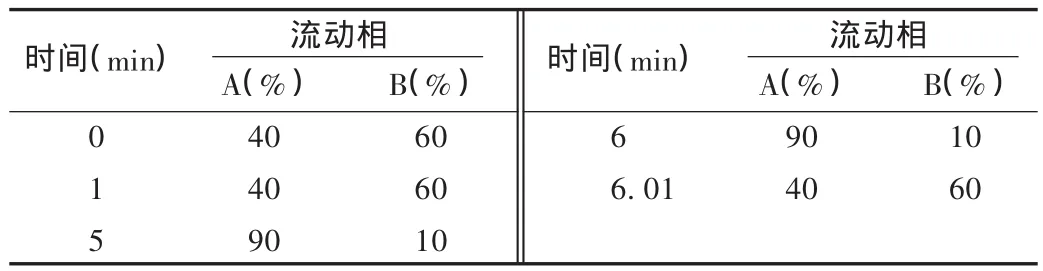
表1 流動相梯度洗脫表
質譜條件:Quattro MicroTM三重四級桿質譜,離子化模式為ESI+,檢測方式為多反應監測(MRM);毛細管電壓3 kV;脫溶劑氣溫度350℃,脫溶劑氣流量600 L/h;錐孔反吹氣30 L/h。錐孔電壓和碰撞能量見表2。
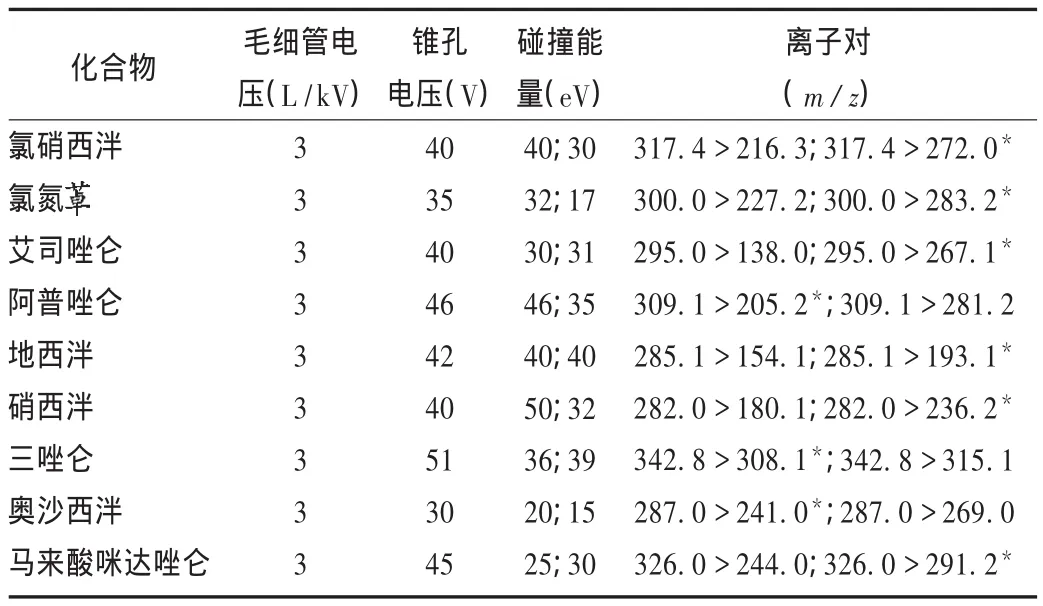
表2 質譜參數
2.2 質譜分析及定性、定量
以電噴霧離子化方式對9種藥物的標準溶液進行MS掃描,9種化合物均形成[M+H]+準分子離子峰,以此作為母離子,進行碎片掃描,選擇豐度較強的2個子離子。經試驗,9種化合物均在正離子檢測方式下具有較高的靈敏度,因此采用正離子檢測的方式進行UPLC-MS/MS分析。定性鑒別時,以化合物的保留時間及2組離子對進行確認。定量測定時以定量離子對的響應做外標法進行定量分析。9種化合物的總離子流圖(TIC)見圖1,二級質譜圖見圖2。
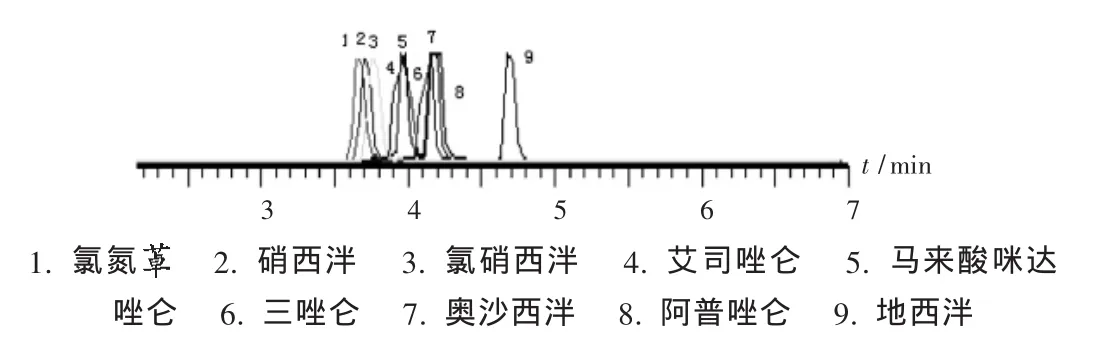
圖1 9種化合物的總離子流圖
2.3 溶液制備
混合標準貯備液:精密稱取經60℃減壓干燥的9種藥物的對照品適量,用甲醇溶解并制成每mL含有50 μg的標準貯備液并保存于冰箱中。
供試品溶液:對于片劑,取10片,研細;對于膠囊劑,取10粒膠囊,將內容物取出,必要時研磨,將膠囊殼剪碎,與內容物混勻;對于蜜丸,將蜜丸粉碎成細小顆粒;對于顆粒劑,取5袋,研細。稱取處理好的樣品的一次口服用量,精密稱定,置100 mL量瓶中,加甲醇約70 mL,超聲提取20 min,放冷至室溫,加入甲醇稀釋至刻度,搖勻,經過夜沉淀后取上層溶液適量,高速離心(不小于105/min),取上清液用流動相溶液稀釋10~100倍,即可。
2.4 方法學考察
線性關系考察:分別精密吸取2.3項下標準貯備液適量,用甲醇稀釋成 25,50,100,250,500,600,800,1 000 μg/L 的混合標準系列濃度溶液,進行UPLC-MS/MS測定。以化合物定量離子對的峰面積(Y)對相應的質量濃度(X)進行線性回歸,見表3。

圖2 9種化合物的二級質譜圖
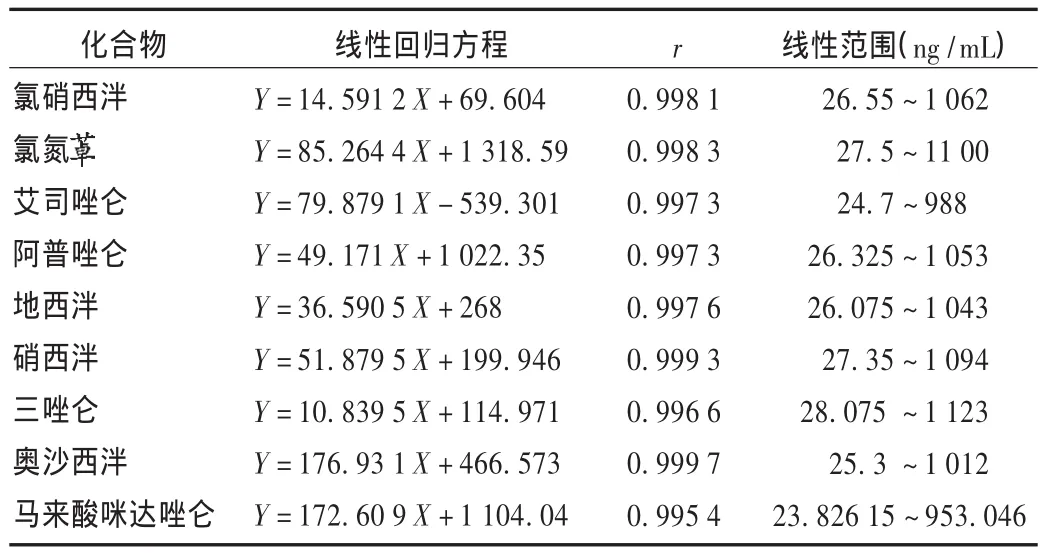
表3 9種化合物線性關系
重復性試驗:精密稱取陽性樣品平行6份,每份1次口服用量,照2.3項下方法制得供試品溶液,進樣5 μL。結果檢出地西泮,其余均未檢出(圖3)。根據地西泮的定量離子峰面積,計算地西泮的平均含量為 0.521 82 mg/g,RSD 為 1.2%(n=6)。
精密度試驗:取300 μg/L加標水平混合標準溶液,在擬訂的UPLC-MS/MS條件下連續測定6次,由氯硝西泮、氯氮、艾司唑侖、阿普唑侖、地西泮、硝西泮、三唑侖、奧沙西泮和馬來酸咪達唑侖定量離子對峰面積計算精密度,RSD分別為4.9%,1.3%,1.8% ,3.6%,2.1%,2.6% ,6.6%,1.3% ,1.5%(n=6)。
檢測限和定量限:取混合對照品貯備液,用甲醇稀釋成一定質量濃度的溶液后測定各化合物的信噪比,依據檢出限為3倍信噪比(S/N=3),定量限為10倍信噪比(S/N=10),分別得出各化合物的檢測限:氯硝西泮1×10-3ng、氯氮0.2 ×10-3ng、艾司唑侖 0.2 ×10-3ng、阿普唑侖 0.3 ×10-3g、地西泮 1.5 ×10-3ng、硝西泮 0.3×10-3ng、三唑侖 1.2 ×10-3ng、奧沙西泮 0.2×10-3ng 和馬來酸咪達唑侖 0.2 ×10-3ng。定量限:氯硝西泮 3 ×10-3ng、氯氮卓 0.7 ×10-3ng、艾司唑侖 0.6 ×10-3ng、阿普唑侖 1 ×10-3g、地西泮 5×10-3ng、硝西泮 1×10-3ng、三唑侖 4×10-3ng、奧沙西泮0.6×10-3ng和馬來酸咪達唑侖 0.6 ×10-3ng。
穩定性試驗:取300 μg/L加標水平混合標準溶液,分別在配制后于室溫下放置 0,3,6,12,18,24 h 時測定,根據地西泮、氯硝西泮和艾司唑侖定量離子對峰面積計算精密度,RSD分別為5.1% ,1.5% ,2.0% ,3.9% ,2.2% ,2.9% ,6.6% ,1.8% ,1.7%(n=6)。結果表明,9種化合物在24 h內基本穩定。

圖3 樣品的多反應監測圖
加樣回收試驗:精密稱取陰性樣品9份,分別精密加入混合標準儲備液適量,照2.3方法提取,終質量濃度分別為200,300,500 μg/L,每個質量濃度水平平行試驗3份,根據外標法計算9種化合物的平均回收率。結果見表4。
專屬性試驗:分別取約3 g空白制劑的粉末和添加了檢測限濃度的9種鎮靜催眠藥的相同制劑粉末,按照2.3項下方法制備。所得到的定量離子對多反應監測色譜圖表明,在9種鎮靜催眠藥的出峰位置,空白樣品中無干擾峰出現,該方法具有較好的專屬性。
2.5 樣品檢測
樣品按2.5項下方法制備,在擬訂的UPLC-MS/MS條件下測定,結果11批抽驗樣品其中2批檢出地西泮,其余均無非法添加。
3 討論
由于中藥成分的復雜性,故有必要采用高選擇性的分析手段。本法可快速、準確地對中藥制劑及保健品中非法添加氯硝西泮、氯氮、艾司唑侖、阿普唑侖、地西泮、硝西泮、三唑侖、奧沙西泮和馬來酸咪達唑侖同時進行檢測,專屬性強,9種化合物可達基線分離,分離速度快。
本法建立了鎮靜安神類中成藥及保健品中非法添加9種化合物的專屬檢測方法。試驗考察了乙腈-水和甲醇-水體系為流動相對目標化合物的分離效果,結果表明,這兩種體系為流動相時,目標化合物的質譜信號響應存在差別,甲醇-水體系靈敏度高且噪音小,且安全和經濟性較乙腈-水好,綜合考慮選擇甲醇-水體系為流動相。在建立方法時分別采用等度洗脫和梯度洗脫的方法進行比較,結果梯度洗脫分離速度明顯比等度洗脫快且分離效果好,因此采用0.1%甲酸-甲醇梯度洗脫。
ESI(+)模式下,常采用加入揮發性酸以降低流動相的pH,有助于待測藥物的離子化。在ESI中,流速大意味著同時進入離子源的化合物也增多。需要同時離子化的化合物增多,加劇了待測成分與基質成分在電離過程的競爭,從而使待測成分的響應降低。此外,在低流速下可產生較細的霧滴,而使霧滴的總表面積增加,待測成分與基質成分在表面積的競爭減少,從而有利于產生氣相離子,提高待測成分的信號強度。該試驗中選擇流速為0.3 mL/min,既避免了待測成分因出峰過快而易受雜質峰的干擾,同時又最大程度地縮短了分析時間,滿足了實際應用中復雜中藥樣品的測定。
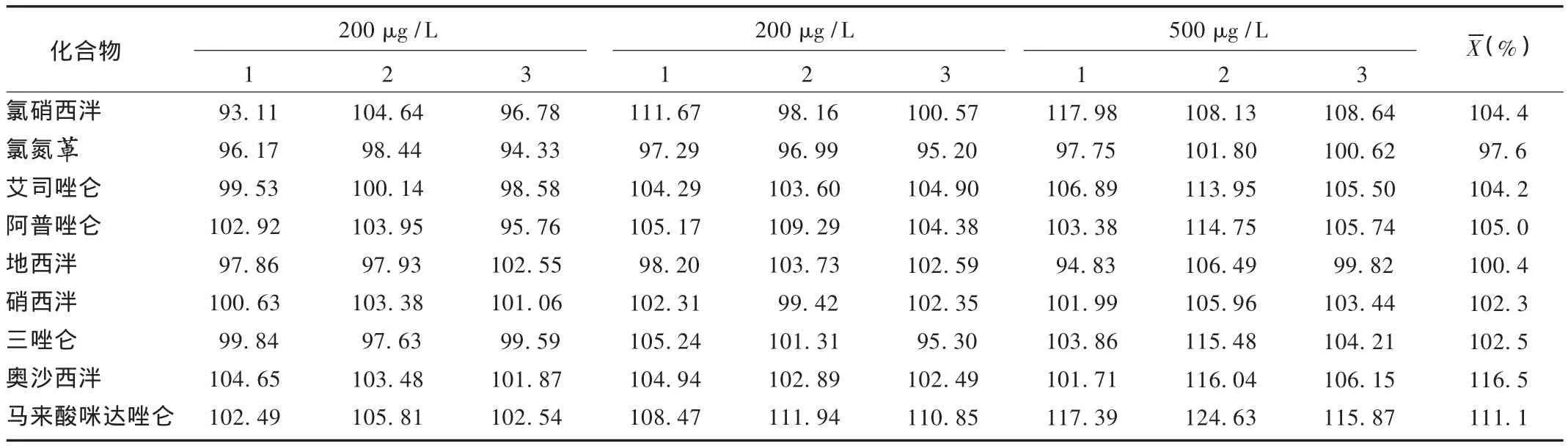
表4 9種化合物的回收率(n=9)
[1]李欣榮,陳安珍,楊 釗.UPLC-MS/MS檢測消腫止痛類中成藥中違禁添加的雙氯芬酸鈉和氨基比林[J].藥物分析雜志,2010,30(7):1 294.
[2]Peng Z,Sharon SY,Peiling H,et al.Simultaneous determination of synthetic phosphodiesterase-5 inhibitors found in a dietary supplement and pre mixed bulk powders for dietary supplements using high-performance liquid chromatography with diode array detection and liquid chromatography-electrosp ray ionization tandem mass spectrometry[J].J Chromatogr A,2006,1 104:113 -122.
猜你喜歡
中國設備工程(2022年12期)2022-07-11 04:33:00
中學生數理化·七年級數學人教版(2021年6期)2021-11-22 07:50:58
中學生數理化·七年級數學人教版(2021年6期)2021-11-22 07:50:58
中學生數理化·七年級數學人教版(2021年6期)2021-11-22 07:50:58
中學生數理化·七年級數學人教版(2020年12期)2021-01-18 06:57:46
中學生數理化·七年級數學人教版(2020年12期)2021-01-18 06:57:46
中學生數理化·七年級數學人教版(2019年9期)2019-11-25 07:34:36
中學生數理化·七年級數學人教版(2019年9期)2019-11-25 07:34:34
中學生數理化·七年級數學人教版(2019年12期)2019-05-21 02:53:50
中學生數理化·七年級數學人教版(2019年12期)2019-05-21 02:53:48
